BackgroundThe St. Nicolas House Analysis (snha) is a new graph estimation method
for detecting extensive interactions among variables. It operates by ranking absolute
bivariate correlation coefficients in descending order thereby creating hierarchic
association chains. The latter characterizes dependence structures of interacting
variables which can be visualized in a corresponding network graph as a chain of
end-to-end connected edges representing direct relationships between the connected
nodes.
ObjectiveThe important advantage of this relatively new approach is that it
produces less false positive edges resulting from indirect or transitive associations
than expected with standard correlation or linear model-based approaches. Here, we aim
to improve the detection of ramifications in graphs by addition of different data
processing layers to snha.
MethodsThe methods include the combinations of the extensions R-squared gaining
(rsg) and linear model check (lmc). The method snha together with these so-called
extensions were benchmarked against default snha and other reference methods available
for the programming language R.
ResultsCombinations of rsg, lmc and bootstrapping improve snha performance
across different network types, albeit at the cost of longer computation time.
ConclusionThe improved accuracy and robustness of network ramification detections
make the integration of combinations of snha extensions a valuable approach for complex
network analysis.
Keywords: St. Nicolas Analysis, snha, network reconstruction, R-squared gaining, linear model check, graph estimation
Conflict of interest: There are no conflicts of interest.
Citation: Chen, S. / Moris, C. / Groth, D. (2024). Improving ramification detection of St. Nicolas House
Analysis. Human Biology and Public Health 1. https://doi.org/10.52905/hbph2024.1.81.
Copyright: This is an open access article distributed under the terms of the Creative Commons Attribution License which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
The St. Nicolas House Analysis R package offers intuitive and fast ways to
interact with your research data while providing a graphical representation of potential
interactions between variables.
Contents
Abbreviations
Shna: St. Nicolaus House Analysis
shna_def: default shna without extensions
rsg: R-squared gaining
lmc: Linear model check
boot: Bootstrapping
lmb: Combination of bootstrapping and lmc
ct: Correlation Thresholding
mb: Meinshausen-Bühlmann
qg: qgraph
bic: Bayesian Information Criterion
ebic: Extended Bayesian Information Criterion
ergm: Exponential Random Graph Models
Sens: Sensitivity
Spec: Specificity
Prec: Precision
Acc: Accuracy
MCC: Matthews’ Correlation Coefficient
BCR: Balanced Classification Rate
NIR: Null Information Rate
TP: True Positives
FP: False Positives
TN: True Negatives
FN: False Negatives
Note: Combinations of methods, such as lmc_rsg, indicate the use of both lmc and rsg.
Introduction
Network reconstruction employs computational techniques to unveil the
intricate web of interactions in systems ranging from biological to social and
technological, elucidating their structure and dynamics. This process is crucial for
grasping how components interact and influence the overall system behavior (Hemelrijk 1990). Commonly, the aim of network
reconstruction is to deduce exclusively causative relationships. For this purpose, numerous
approaches have been proposed. They may be designed for specific applications such as
protein structure prediction (Marks et al. 2011) or
gene expression networks (Logsdon and Mezey 2010).
More general-purpose methods may formulate the task as a feature-selection problem (Huynh-Thu et al. 2010; Meinshausen and Bühlmann 2006), or leverage partial correlation
information to characterize relationships among variables (Hemelrijk 1990).
Here we focus on the St. Nicolas House analyses (snha), first published in
2019 (Groth et al. 2019). The shna takes a
different approach: It searches for so-called association chains. Association chains
incorporate the domino effect of one variable influencing the next one, which itself
influences a third variable on its own, and so on.
shna looks for association chains in the correlation matrix of a given
dataset. Starting with the first variable , the correlations with the other variables or nodes are
ranked in non-increasing manner. Let us say that this results in the correlation coefficient
between and being the greatest, followed by the slightly lower one
between nodes and and an even smaller one between nodes
and ). When ranked hierarchically, we get
|, and therewith also the chain G-O-C-M. We define this as the
forward chain. Next, the sequence is inverted, and the end node M from the forward chain now
constitutes the starting node. We check whether for this backward chain M-C-O-G the same
hierarchic order of magnitude is represented in the correlation matrix:
|. These sequences of correlation coefficients, that are
characterized by descending order when starting from either end, are named “association
chains”. Association chains can be translated into network graphs. At first view, the whole
procedure sounds trivial, but it opens the possibility of immediately visualizing extensive
interacting variables in an explorative manner (Hermanussen et al. 2021).
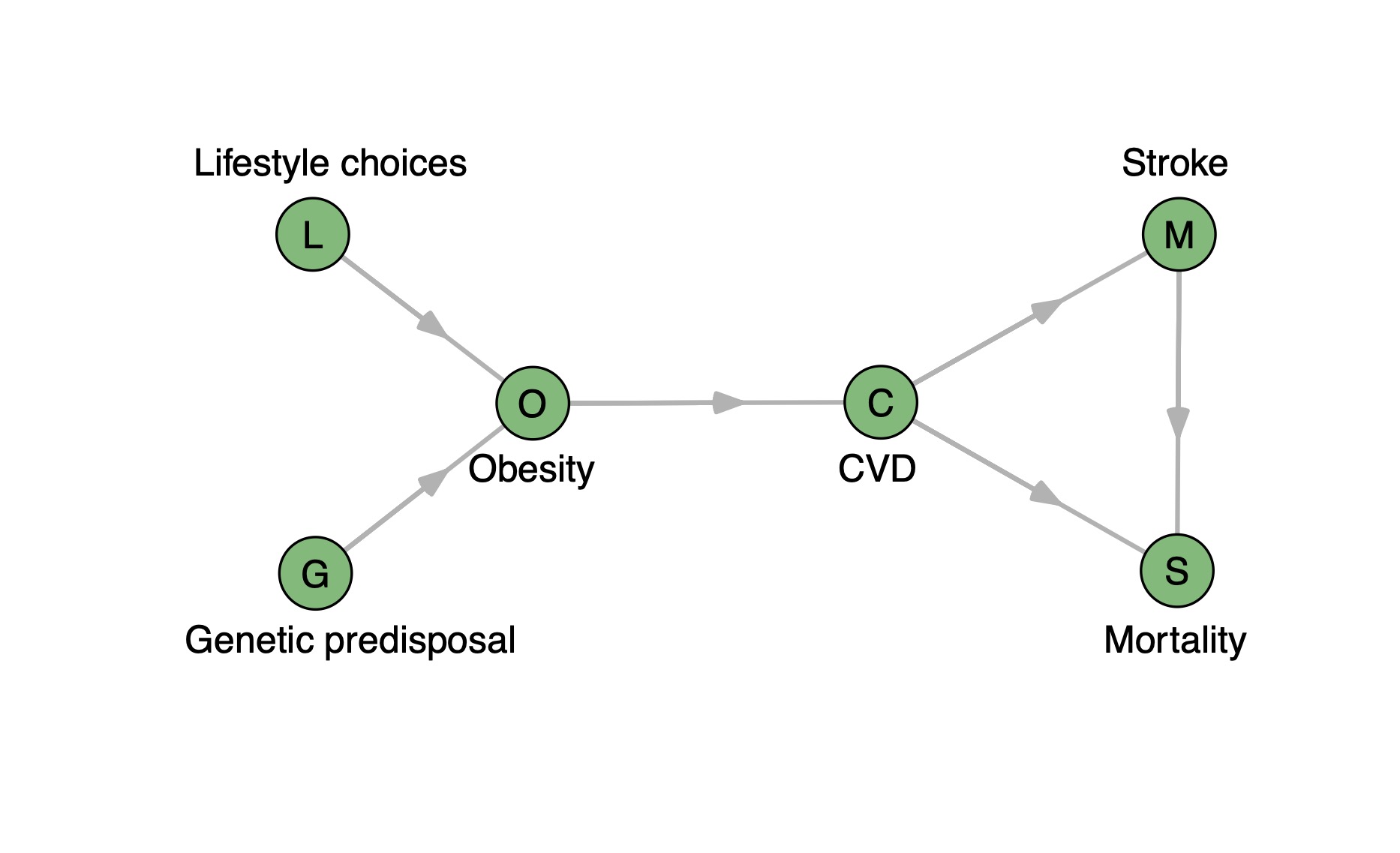
To illustrate the approach and simultaneously a weakness of shna, the
following directed graph is introduced (Figure 1).
There are two input variables depicted as nodes, genetic predisposal and lifestyle choices,
that influence the target variable obesity. Obesity heightens the risk of cardiovascular
diseases (CVD), and CVD in turn increases mortality among the affected population.
Additionally, CVD is a risk factor for stroke, hence the graph also includes a directed edge
from the node CVD to the node stroke. Stroke, on the other hand, greatly increases
mortality, whose node ends up as the sink node. Note that the triangle on the right is not a
cycle due to the directionality of the edges. Therefore, the resulting network is a typical
directed acyclic graph (DAG).
Figure 1 A directed so-called Werner graph visualizing the obesity-mortality example. Nodes
represent variables and edges direct interactions between them. CVD: cardiovascular
diseases.
Due to its approach to find linear association chains, snha struggles with
the detection of ramifications. When given a dataset or correlation matrix based on the
example graph, it is most likely to miss one of the outgoing edges from CVD. However, the
potentially missing relationship describes a causative relationship, thus it is too
important to ignore (and tolerate) its absence. Accordingly, the aim of this study is to
improve the capability of snha to detect ramifications in the underlying interdependencies
of the input data with only little increase in runtime.
Materials and methods
Software
Programming was done exclusively with the language R for this project
(R Core Team 2022). Additionally, the R
packages used include huge (Jiang
et al. 2021), qgraph (Epskamp
et al. 2012), ergm (Krivitsky
et al. 2023), snha (Groth
2023), mcgraph and asg (Groth 2022), of which huge, qgraph and
ergm can be considered references of network reconstruction. Synthetic
data was generated with functions from the mcgraph package (Novine et al. 2022). A public version of
mcgraph can be found within the snha package called
mgraph.
Graphs used
Barabasi graphs, also known as Barabasi–Albert (BA) models, are a type of
scale-free network characterized by preferential attachment mechanism, where new nodes
are more likely to connect to existing nodes with higher degrees (Barabasi and Albert 1999).
Density in the context of graphs refers to the ratio of the number of
actual edges in the graph to the maximum possible number of edges among all nodes. It
provides a measure of how closely knit the connections are in the network, with higher
density indicating more connections per node and lower density signifying fewer
connections per node (Diestel 2017).
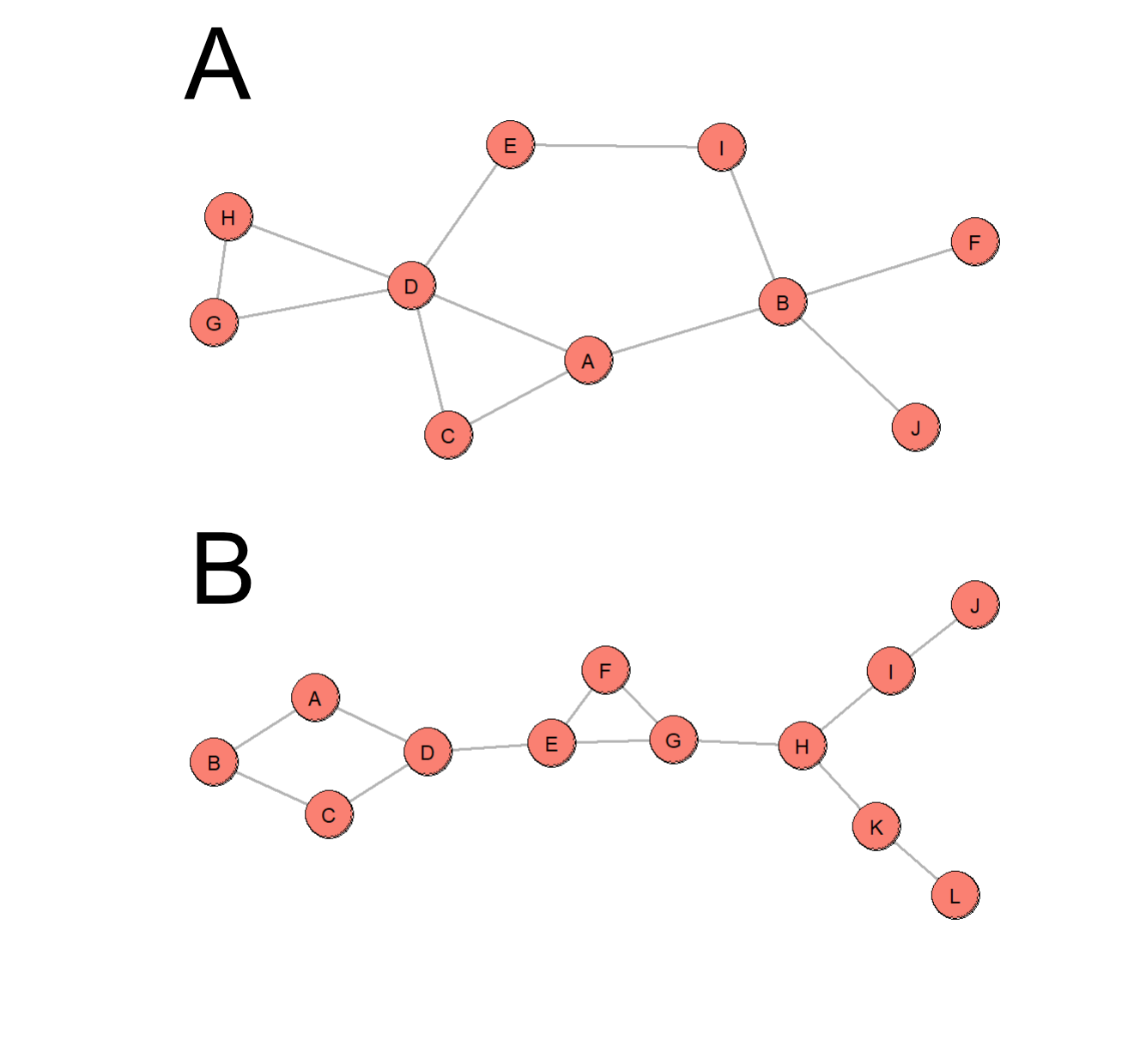
The graphs analyzed include Barabasi graphs (Barabasi and Albert 1999) of density 1.5 denoted as M1.5, Werner (W)
and Wernerextended (WX), an extended version of W, graphs. WX has an additional
diamond-like structure and a longer branching path compared to W. We included the M1.5
as it was suggested to be a more reasonable representation of biological networks. These
M1.5 graphs were made in a way that either adds 1 or 2 edges at each step of the
Barabasi graph generation. We chose to only include Barabasi graphs with 20 nodes. The
Barabasi graph in Figure 2 has 10 nodes and WX is
shown with 12 nodes as visual representations of the graphs used in our tests.
Figure 2 A: Barabasi graph with 10 nodes of densities 1.5 B: Wernerextended graph with
12 nodes.
Data generation and benchmarking
In order to benchmark different graph prediction methods, we decided to
create synthetic data using Novine’s Monte Carlo implementation (Novine et al. 2022), the function is called mgraph.graph2data from
the public snha package. Some testing was done with
huge data generation. Synthetic data built upon a known graph that we
gave as an input allows us to easily compare the predicted graph against the original
one.
The huge package contains implementations of different
methods for network reconstruction, of which we chose correlation thresholding (ct),
Meinshausen-Bühlmann (mb) (Meinshausen and Bühlmann
2006) covariance selection and graphical least absolute shrinkage and selection
operator (glasso) (Friedman et al. 2008). As in
most cases the true underlying graph structure is unknown, we opted to use the automatic
lambda selection function for the huge methods. Furthermore, glasso and
lasso implementations of qgraph were chosen with either Bayesian
Information Criterion (bic) or extended Bayesian Information Criterion (ebic) (Epskamp et al. 2012).
To complement these approaches and provide a comparative methodological
framework, we incorporated Exponential Random Graph Models (ergm) from
the ergm package (Krivitsky et al.
2023). The ergms are particularly well-suited for modeling complex network data
through specified probability distributions over graphs, thus offering a robust
alternative to regularized regression techniques used in glasso and lasso (Krivitsky et al. 2023). This allows us to contrast
the performance of traditional regression-based network reconstruction techniques with
that of stochastic models tailored for social network analysis and other fields where
the underlying network dynamics are inherently probabilistic.
The benchmarking procedure was generally structured as follows:
Initially, a graph was constructed utilizing the mgraph.new function from the asg or the
snha package. The adjacency matrix derived from this graph served as a blueprint for the
generation of simulated data. This simulation aimed to mimic real data as accurately as
possible by maintaining the graph’s interdependencies in a balanced manner and
incorporating random noise, ensuring that the inherent connections were neither
exaggerated nor understated (Novine et al.
2022). The result was a synthetic dataset wherein the quantity of variables
matched the number of nodes in the graph. This dataset, or its correlation matrix if
necessary, was then used as input for the graph estimation functions, which produced the
adjacency matrix of the estimated graph. The final step involved evaluating each
method’s performance by contrasting the estimated graph with the original one.
Accuracy metrics used
There has long been disagreement in the scientific community about which
accuracy standards should be used (Bekkar et al.
2013), most of these metrics are based on ratios of true positives (TP), true
negatives (TN), false positives (FP) and false negatives (FN). In our case, they
correspond to correct and incorrect numbers of predicted edges or non-edges.
The sensitivity (Sens) is defined as the ratio of true positives to the
sum of true positives and false negatives.
The specificity (Spec) is defined as the ratio of true negatives to the
sum of true negatives and false positives.
Precision (Prec) is calculated as the ratio of true positives to the sum
of the true positives and false positives.
The Matthews Correlation Coefficient (MCC) (Chicco and Jurman 2020) is calculated as follows:
As the MCC does not fully capture the disproportionate gain of either
Sens or Spec compared to the other, we included the Balanced Classification Rate (BCR)
(García et al. 2009) and No Information Rate
(NIR) (Bicego and Mensi 2023). BCR is the
average of Sens and Spec, so a large gain in Sens with a small loss in Spec will still
yield a better BCR.
One could say that NIR is something like an estimate of the most common
class without any information and serves as a baseline metric. If for example 70% of the
data is class A and 30% is class B, then estimates of class A would every time yield an
accuracy of 70%. The NIR, in this context, represents this baseline accuracy. Therefore,
the NIR is used to compare model accuracy against a baseline. If the accuracy (Acc)
surpasses the NIR greatly, it has significant predictive power. Therefore, Acc and NIR
should be viewed together. The Acc is calculated as the ratio of the sum of true
positives and true negatives to the total number of cases.
R squared gaining
The extension called R-squared gaining (rsg) follows the concept of
aiming at improving a specific scoring measure. In rsg, the aim is to maximize the gain
in adjusted R-squared values obtained with the lm function in the stats
package of base R. The R-squared value represents the proportion of variance of the
response variable, here the target node, that is explained by the predictor variable(s),
or the source node(s). The normal R-squared statistic refers to the sample and the
adjusted R-squared value to an estimate in the population (Miles 2005).
For every node i in the graph, rsg searches for candidate nodes with
which a new edge appears meaningful, with respect to their original correlation
coefficients and p-values, which are output by default snha. Then, a linear regression
model was fitted with the node i as response variable and the already connected node(s)
as predictor variable(s). A second model included the candidate node as additional
predictor variable. If the second model resulted in an adjusted R-squared value higher
than the first model, and if the difference was over a specified threshold, then the
edge between the node i and the candidate node was added to the graph. This threshold
was the only parameter in this extension and consequently, it was tested for a range of
values.
Linear model check
Another method, linear model check (lmc), has been implemented for this
project. After obtaining a predicted graph, for example via the default snha, using
linear models to check the obtained snha graph to the data, edges will be removed by lmc
if they do not add more than a set threshold of the adjusted R-squared value. We set the
default threshold to two percent.
Combinations
The novel approach in this project was the combination of extensions of
snha, as weaknesses and strengths of each method may balance each other out.
Multiple combinations of extensions were tested. A combination of rsg and
lmc was thus denoted as rsg_lmc, which initially applied rsg followed by lmc and lmc_rsg
follows the same naming-logic in inversed order, meaning that we first applied lmc and
then rsg. That means that lmb combined lmc and bootstrapping (boot) and boot_lmc_rsg
combined boot, lmc and rsg.
Afterwards, pairwise t-tests were conducted to compare whether the
extensions significantly outperformed or underperformed compared to the default snha.
These t-tests were performed in a paired manner with each individually created graph at
each run. This enhanced the comparability as a particularly poor prediction on a “harder
graph”, e.g. containing diamond and triangle patterns, which are more challenging to
resolve, will not be compared against a good prediction of a different method on an
“easy graph”. Due to the nature of the Barabasi graph generation, some randomness can be
expected, which enabled the possibility of creating graphs of varying difficulty in
regard to the ramifications.
Results
Overview
Interestingly using huge data generation yielded better
results for the methods from the huge package. Both ct and mb benefited
heavily from using the synthetic data generated by the huge function,
whereas performance was worse when using our generated data. All other methods did not
show this trend, thus we decided to only include results using Novine’s MC data generation
(Novine et al. 2022).
The new extensions yielded better results, especially combinations of rsg
and lmc in either orientation. Using both subsequently generally produced better results
than each method in isolation when regarding the MCC. Compared to the reference methods of
the huge, qgraph and ergm packages, our methods seem to
consistently strike a better balance between Sens and Spec, since neither of those two
metrics dips below 0.5 for any combination of snha extensions. A comprehensive overview
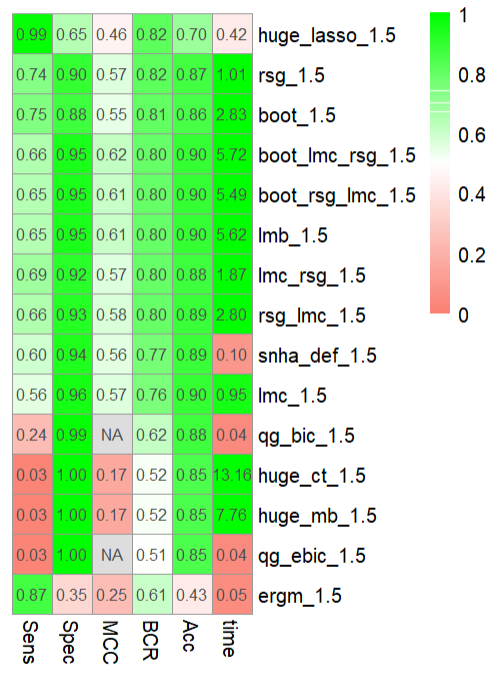
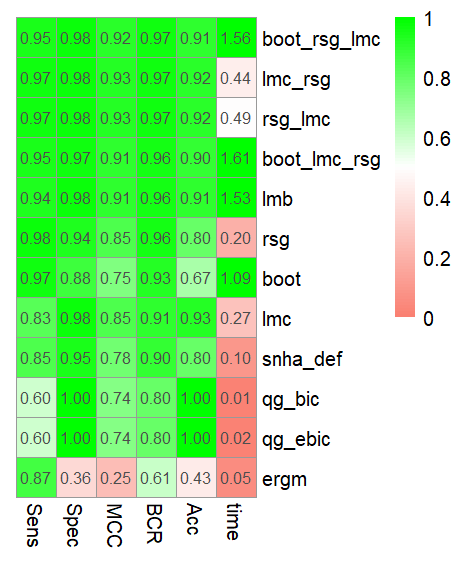
can be seen in Figure 3.
Figure 3 Heatmap showing the means of 10 runs of M1.5 with 20 nodes for Sensitivity
(Sens), Specificity (Spec), Matthews correlation coefficient (MCC), Balanced
Classification rate (BCR), time in seconds and accuracy (Acc) for network
reconstruction methods huge correlation threshold (huge_ct), huge lasso (huge_lasso),
huge Meinshausen-Bühlmann (huge_mb) , qgraph glasso with bic (qg_bic), qgraph glasso
with ebic (qg_ebic), exponential random graph models (ergm), St. Nicolas House
Analysis (snha) and its extensions Linear Model Check(lmc), R-squared Gaining(rsg) and
bootstrapping (boot).The Null Information Rate (NIR) to compare against is 0.85.
Matthews correlation coefficient and balanced classification rate
Barabasi M1.5
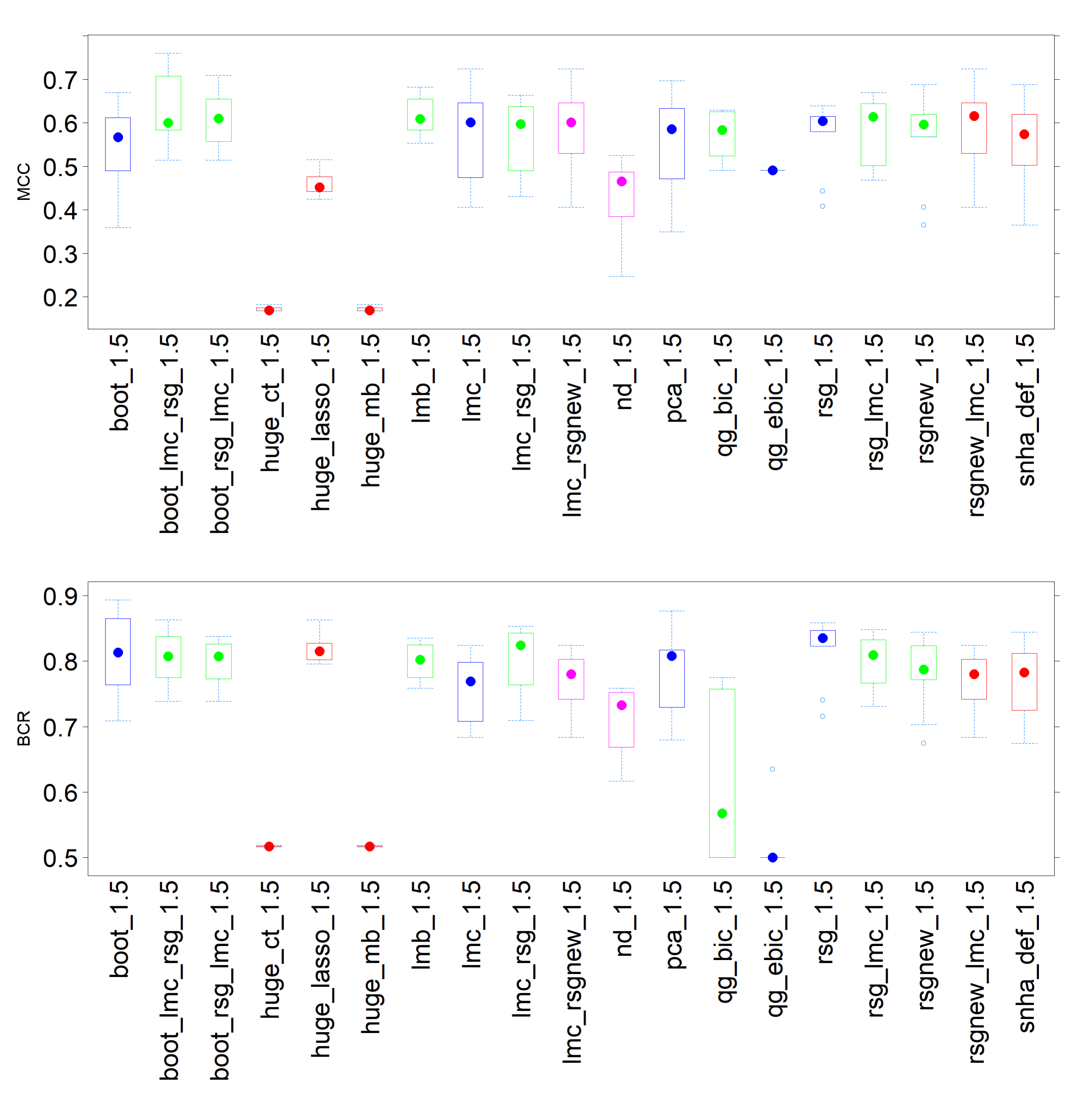
Figure 4 shows a box and whiskers
plot for the MCC and the BCR. Within the scope of MCC, it is observed that the boxes are
generally quite elongated for all methods. This observation implies a lack of
consistency across the performance of all methods.
When considering the BCR, the subpar performance of the
huge methods, specifically mb and ct, becomes apparent. Despite their
high Spec, their Sens is low, which results in a diminished BCR. Conversely, the
combinations of lmc and rsg, along with boot, emerge as some of the top performers in
terms of BCR. This suggests that these combinations are more adept at striking a balance
between Sens and Spec. Additionally, as visible in Figure 1, the MCC was not always calculable for qgraph lasso with bic or
ebic as selection criterion.
For M1.5, the range was between 0.8 and 0.9. In all cases, the default
shna fell short when compared to boot, lmc, rsg, and combinations of these three.
Figure 4 Box and whiskers plot of Matthews correlation coefficient (MCC) and Balanced
Classification rate (BCR) for M1.5 using various network reconstruction methods.
Different methods are colorcoded as follows: huge functions are red, qgraph
functions are magenta, singular extensions of St. Nicolas House Analysis (snha) and
default snha are blue and combinations of extensions are
green.
The paired t-tests for BCR, presented in Tab. 1, reveal that most combination methods incorporating lmc
significantly outperform the default snha. In contrast, qgraph lasso with bic and ebic,
as well as huge ct and mb, perform significantly worse than the default snha. It becomes
evident that combinations involving at least either boot or lmc tend to perform better
than the default snha.
Table 1 Paired t.tests for Balanced Classification rate(BCR) in M1.5 with 20 nodes
comparing network reconstruction methods against default St. Nicolas House Analysis
(snha_def). These include snha with bootstrapping (boot), Linear Model Check (lmc),
lmc and boot (lmb), huge correlation threshold (huge_ct), huge lasso (huge_lasso),
huge Meinshausen-Bühlmann (huge_mb) and qgraph lasso with either bic or ebic (qg_bic
and qg_ebic).
estimate
conf.low
conf.high
p.value
snha_def_vs lmc_1.5
0.012
-0.008
0.032
0.210
snha_def_vs lmb_1.5
-0.029
-0.054
-0.005
0.023
snha_def_vs boot_1.5
-0.043
-0.066
-0.020
0.002
snha_def_vs rsg_1.5
-0.046
-0.068
-0.025
0.001
snha_def_vs rsg_lmc_1.5
-0.027
-0.060
0.006
0.102
snha_def_vs
boot_rsg_lmc_1.5
-0.028
-0.049
-0.006
0.016
snha_def_vs lmc_rsg_1.5
-0.030
-0.068
0.007
0.096
snha_def_vs
boot_lmc_rsg_1.5
-0.033
-0.056
-0.010
0.010
snha_def_vs huge_ct_1.5
0.253
0.215
0.292
0.000
snha_def_vs
huge_lasso_1.5
-0.047
-0.088
-0.006
0.028
snha_def_vs huge_mb_1.5
0.253
0.215
0.292
0.000
snha_def_vs qg_ebic_1.5
0.257
0.210
0.305
0.000
snha_def_vs qg_bic_1.5
0.155
0.046
0.264
0.011
Wernerextended
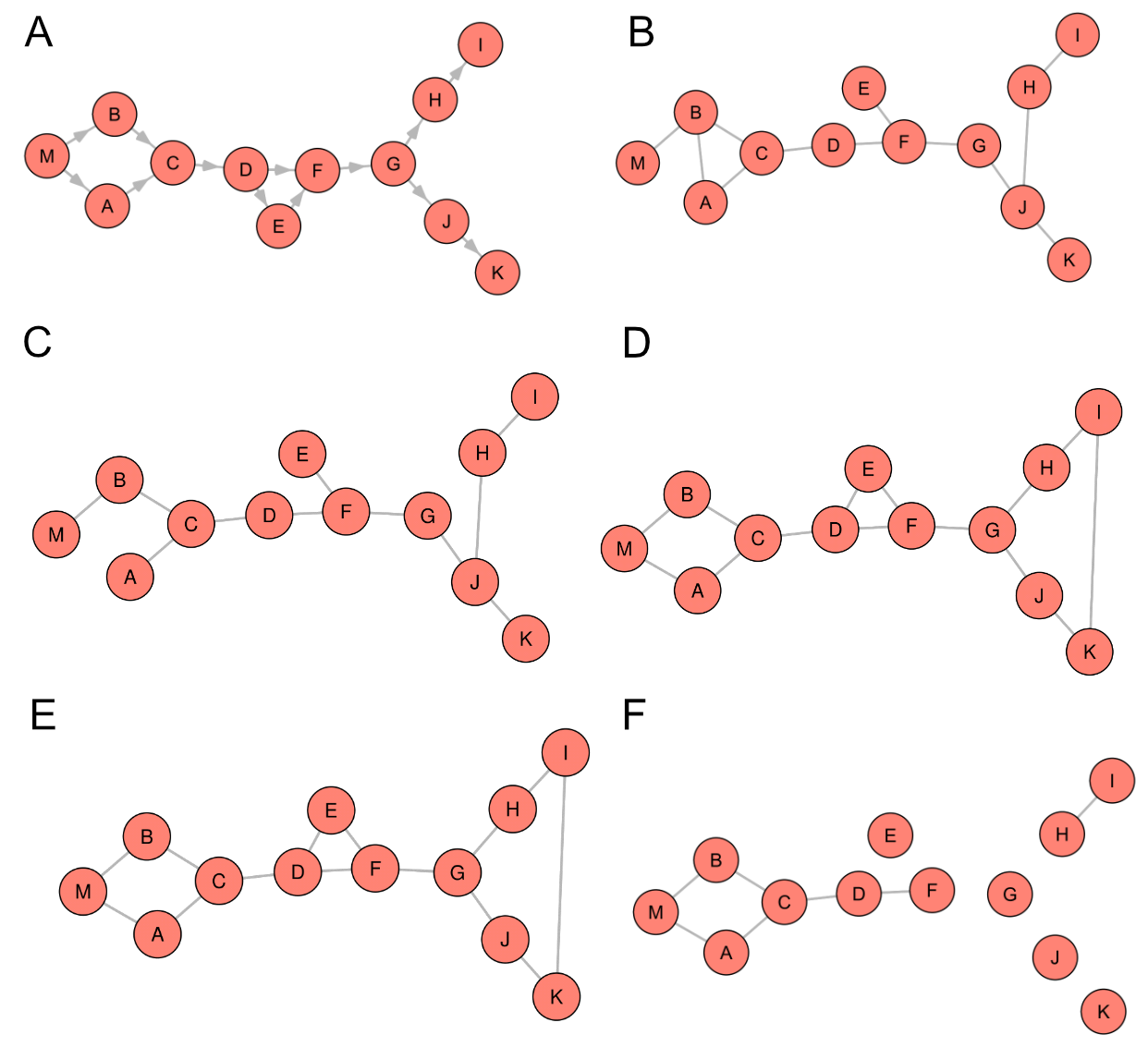
Figure 5 provides a visual
representation of the predicted Wernerextended (WX) graphs using default snha, lmc,
lmc_rsg, lmb_rsg and qgraph lasso. The results observed in the Barabasi
graphs are mirrored in these graphs. Specifically, qgraph predicts fewer edges, but all
of them are correctly identified. In contrast, our methods predict a larger number of
edges, some of which are incorrectly predicted. The number of mistakes decreases with
the addition of more extensions, but an edge at the end of the branching path between
nodes I and K is added for both lmc_rsg and lmb_rsg.
The challenges previously encountered with the triad consisting of nodes
D, E, and F have been addressed by the combination of extensions. The branching path
originating from node G can also be correctly identified by these combination methods.
Interestingly, with bootstrap, lmb_rsg can also reconstruct the diamond-like structure,
correctly assigning both edges to nodes A and B from node M.
qgraph, on the other hand, can solve the diamond-like
structure and the path from node C to nodes D and F. However, it fails to correctly
identify the triad and the branching path.
Figure 5 Applying different graph estimation methods on Wernerextended (WX) graph. A:
original WX, B: default St. Nicolas House Analysis (snha), C: Linear Model Check
(lmc), D: lmc and R-squared Gaining (rsg), E: Linear Model Check with Bootstrapping
(lmb) and rsg, F: qgraph lasso
As evident in Figure 6, default
snha and qgraph glasso are the least effective at predicting the WX graph based on the
BCR and MCC metrics. However, they are the fastest methods in terms of computational
time. The combination methods, on the other hand, perform well, with boot_lmc_rsg,
lmc_rsg, rsg_lmc and boot_lmc_rsg emerging as the top performers. These methods maintain
a high Sens while achieving a very high Spec. While their Spec, at 0.98, is slightly
lower than qgraph’s perfect score of 1.00, their Sens, ranging from 0.95 to 0.97, is
significantly higher than qgraph lasso’s Sens of 0.6.
The combination methods exhibit better Sens compared to default snha and
are close in terms of Spec, indicating that they detected more edges. This observation,
in conjunction with Figure 5, confirms that the
ramifications that were discovered indeed correspond to the additional edges
detected.
Figure 6 Heatmap showing the means of 10 runs of M1.5 with 20 nodes for Sensitivity
(Sens), Specificity (Spec), Matthews correlation coefficient (MCC), Balanced
Classification rate (BCR),time in seconds and Accuracy (Acc) for network
reconstruction methods huge correlation threshold (huge_ct), huge lasso
(huge_lasso), huge Meinshausen-Bühlmann (huge_mb) , qgraph glasso with bic (qg_bic),
qgraph glasso with ebic (qg_ebic), exponential random graph models (ergm) , St.
Nicolas House Analysis (snha) and its extensions Linear Model Check(lmc), R-squared
Gaining(rsg) and bootstrapping (boot).The Null Information Rate (NIR) to compare
against is 0.80.
Time
All bootstrap variants exhibit a slowdown, with their computational times
being at least an order of magnitude greater than the others. The huge methods utilizing
the star selection criterion also demonstrate a similar behavior. While rsg_lmc and
lmc_rsg perform within an acceptable timeframe, they are noticeably slower than the
default snha.
The default snha and singular extensions require a second or less of
computational time. Combinations of rsg and lmc take less than 3 seconds, but adding boot
to the mix increases the computational time to between 5 and 6 seconds for M1.5.
For M1.5 graphs with 20 nodes, the default snha takes approximately 0.095
seconds, while the boot_lmc_rsg combination requires an average of 5.72 seconds. This
equates to nearly a 60-fold difference in computational time.
Discussion
The original intent of snha was to provide a quick overview of one’s data,
assisting in hypothesis generation and uncovering intriguing connections between variables.
Due to this project and the expansion of extensions for snha, various aspects have changed,
such as the computational time requirement.
Given the graph sizes we are dealing with, the additional time investment
required for improved results is acceptable. While the computational time might become a
concern for larger graph sizes, we anticipate that the number of variables we need to
predict will not be so large as to make computational time a significant issue. To put it
into perspective, if we were to equate the amount of synthetic data we created for 20 nodes
(each with 200 data points) to real-world data, it would be roughly equivalent to the sample
sizes in clinical placebo-controlled trials, which typically measure fewer than 20
parameters across several dozens to hundreds of patients, an example would be trials for
supplements which typically have less than 50 participants (Wu et al. 2022).
As demonstrated in previous works by Bodenberger (Bodenberger) and Moris (Moris
2023), both bootstrapping and rsg outperform the default snha. Not surprisingly,
the combination of bootstrapping with lmc and rsg consistently yielded the best results in
Barabasi graphs of varying densities and in the WX graph.
Our results suggest that rsg may be prone to detecting too many edges, as
indicated by its high sensitivity and slightly lower specificity. Similarly, some methods
have been noted for their propensity to infer a higher number of connections, that may not
reflect true biological interactions. For example, Bayesian network approaches are powerful
for inferring causal networks but can sometimes result in the construction of overly complex
networks, especially when dealing with extensive systems genetics data. The complexity of
diseases like Alzheimer’s or type 2 diabetes often requires careful consideration of
gene-to-gene and gene-to-environment interactions (Tasaki
et al. 2015). Methods developed to simulate co-expression data and evaluate the
performance of various network reconstruction models may also exhibit tendencies to generate
networks with high edge counts, especially under conditions of varying noise levels and
sample sizes. This overestimation can be attributed to the assumptions inherent in the
generative models used for simulation and the performance metrics applied to evaluate method
outcomes (Tasaki et al. 2015).
Conversely, lmc appears to exhibit the opposite trend. Interestingly, when
combined, they seem to balance each other out, while also being faster than any variant of
the bootstrap extension.
The less elongated boxes of BCR compared to those of MCC indicate less
variability in the BCR scores across different methods. This could suggest that BCR might be
a more effective metric overall, providing a more consistent measure of performance across
different network reconstruction methods. This consistency could make BCR a more reliable
metric for comparing the effectiveness of different methods, particularly when the goal is
to achieve a balance between sensitivity and specificity.
“Stability Indicators in Network Reconstruction” discusses the variability of
network reconstruction methods which might imply similar observations about the stability
and reliability of metrics (Filosi et al.
2014).
In the context of the MCC scores, it is worth noting that the huge lasso
method shows a peculiar trend of a slight increase in MCC as the graphs become denser. This
could suggest a better performance of huge lasso in denser graphs. This observation aligns
with expectations from network reconstruction methodologies, where denser connectivity
patterns may provide more data points for lasso-based methods to leverage, potentially
leading to more accurate edge prediction. For comprehensive insights into lasso
methodologies and their applications in dense graph scenarios, the works by Tibshirani
(Tibshirani 1996) on regression shrinkage and
selection via the lasso and Meinshausen and Bühlmann (Meinshausen and Bühlmann 2006) on high-dimensional graphs and variable selection
with the lasso provide foundational understanding.
Another unique behavior observed only in huge mb, ct, and lasso methods is
their tendency to either have a sensitivity or specificity very close to 0 or 1. This
indicates that these methods either predict the existence of all possible edges within a
given graph or predict a single edge with a high degree of certainty. Chen and Mar (Chen and Mar 2018) bring insight into the sensitivity
and specificity trade-offs in network reconstruction and discuss evaluation metrics in the
context of gene regulatory network inference. Similar to our testing, Chen and Mar (Chen and Mar 2018) obtained strongly varying networks
for different reconstruction methods.
It is notable that the original publications on huge methods may not
explicitly discuss the tendency to polarize sensitivity or specificity (Zhao et al. 2020). This gap suggests an opportunity for
future research to explore the conditions under which huge methods, particularly in the
context of mb, ct, and lasso variants, lean towards these extremes.
Efforts to optimize the current rsg extension have so far resulted in a
slight decrease in computational time at the expense of performance. Potential improvements
to rsg, aimed at reducing the number of falsely predicted edges, could include changing the
current threshold to selection criteria that disproportionately penalize the inclusion of
more parameters, such as the Akaike Information Criterion (AIC) (Bozdogan 1987).
5. Conclusion
The inherent ability of the default snha to identify long chains has been
preserved in our combination approaches. Therefore, we conclude that our recent combination
methods have led to improvements in detecting ramifications and enhancing the overall
prediction quality of graphs. This suggests that these combination methods are not only
capable of maintaining the strengths of the default snha, but also of addressing its
limitations, thereby providing a more comprehensive and accurate network reconstruction.
Acknowledgements
I would like to thank Detlef Groth for his guidance during this project. My
sincere thanks also go to Cedríc Morris for his extensive preliminary work which provided a
solid foundation for this project. I am grateful to Masiar Novine for stimulating
discussions and inspiring ideas. I would also like to thank the International Student Summer
School in Gülpe at the University of Potsdam for their support and to the Summerschool KoUP
Funding, without which this work would not have been possible. A special thanks goes to Mari
Delor who provided me with some data to test our methods.
Bekkar, M./Djemaa, H. K./Alitouche, T. A. (2013).
Evaluation measures for models assessment over imbalanced data sets. Journal of
Information Engineering and Applications 3, 27–38. Available online at https://api.semanticscholar.org/CorpusID:52267786.
Bicego, M./Mensi, A. (2023). Null/No Information
Rate (NIR): a statistical test to assess if a classification accuracy is significant for
a given problem, 2023. Available online at http://arxiv.org/pdf/2306.06140.
Bodenberger, B. Improved network reconstruction
using resampling methods. Project work thesis at University of Potsdam.
Potsdam.
Bozdogan, H. (1987). Model selection and Akaike’s
Information Criterion (AIC): The general theory and its analytical extensions.
Psychometrika 52 (3), 345–370. https://doi.org/10.1007/BF02294361.
Chen, S./Mar, J. C. (2018). Evaluating methods of
inferring gene regulatory networks highlights their lack of performance for single cell
gene expression data. BMC Bioinformatics 19 (1), 232. https://doi.org/10.1186/s12859-018-2217-z.
Chicco, D./Jurman, G. (2020). The advantages of
the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary
classification evaluation. BMC Genomics 21 (1), 6. https://doi.org/10.1186/s12864-019-6413-7.
Epskamp, S./Cramer, A. O./Waldorp, L.
J./Schmittmann, V. D./Borsboom, D. (2012). qgraph: Network visualizations of
relationships in psychometric data. Journal of Statistical Software 48 (4),
1–18.
Filosi, M./Visintainer, R./Riccadonna, S./Jurman,
G./Furlanello, C. (2014). Stability indicators in network reconstruction. PLOS ONE 9
(2), 1–24. https://doi.org/10.1371/journal.pone.0089815.
Friedman, J./Hastie, T./Tibshirani, R. (2008).
Sparse inverse covariance estimation with the graphical lasso. Biostatistics 9 (3),
432–441. https://doi.org/10.1093/biostatistics/kxm045.
García, V./Mollineda, R. A./Sánchez, J. S. (2009).
Index of balanced accuracy: A performance measure for skewed class distributions. In:
Helder Araujo/Ana Maria Mendonça/Armando J. Pinho et al. (Eds.). Pattern recognition and
image analysis. Berlin, Heidelberg, Springer Berlin Heidelberg,
441–448.
Groth, D. (2022). Asg: Package for generating
correlation networks based on association chains. Available online at https://github.com/mittelmark/snha-gui.
Groth, D./Scheffler, C./Hermanussen, M. (2019).
Body height in stunted Indonesian children depends directly on parental education and
not via a nutrition mediated pathway - Evidence from tracing association chains by St.
Nicolas House Analysis. Anthropol Anz 76 (5), 445–451. https://doi.org/10.1127/anthranz/2019/1027.
Hemelrijk, C. K. (1990). A matrix partial
correlation test used in investigations of reciprocity and other social interaction
patterns at group level. Journal of Theoretical Biology 143 (3), 405–420. https://doi.org/10.1016/S0022-5193(05)80036-0.
Hermanussen, M./Aßmann, C./Groth, D. (2021). Chain
reversion for detecting associations in interacting variables—St. Nicolas House
Analysis. International Journal of Environmental Research and Public Health 18 (4),
1741. Available online at https://www.mdpi.com/1660-4601/18/4/1741.
Huynh-Thu, V. A./Irrthum, A./Wehenkel, L./Geurts,
P. (2010). Inferring regulatory networks from expression data using tree-based methods.
PLOS ONE 5 (9), 1–10. https://doi.org/10.1371/journal.pone.0012776.
Krivitsky, P. N./Hunter, D. R./Morris, M./Klumb,
C. (2023). ergm 4: New features for analyzing exponential-family random graph models.
Journal of Statistical Software 105 (6), 1–44. https://doi.org/10.18637/jss.v105.i06.
Logsdon, B. A./Mezey, J. (2010). Gene expression
network reconstruction by convex feature selection when incorporating genetic
perturbations. PLOS Computational Biology 6 (12), 1–13. https://doi.org/10.1371/journal.pcbi.1001014.
Marks, D. S./Colwell, L. J./Sheridan, R./Hopf, T.
A./Pagnani, A./Zecchina, R./Sander, C. (2011). Protein 3D structure computed from
evolutionary sequence variation. PLOS ONE 6 (12), 1–20. https://doi.org/10.1371/journal.pone.0028766.
Meinshausen, N./Bühlmann, P. (2006).
High-dimensional graphs and variable selection with the Lasso. The Annals of Statistics
34 (3), 1436–1462. https://doi.org/10.1214/009053606000000281.
Miles, J. (2005). R-squared, adjusted R-squared.
In: Brian Everitt/David Howell (Eds.). Encyclopedia of statistics in behavioral science.
John Wiley & Sons, Ltd.
Moris, C. (2023). Improving ramification detection
of St. Nicolas House Analysis. Project work thesis at University of
Potsdam.
Novine, M./Mattsson, C. C./Groth, D. (2022).
Network reconstruction based on synthetic data generated by a Monte Carlo approach.
Human Biology and Public Health 3. https://doi.org/10.52905/hbph2021.3.26.
R Core Team (2022). R: A Language and Environment
for Statistical Computing. Vienna, Austria. Available online at https://www.R-project.org/.
Tasaki, S./Sauerwine, B./Hoff, B./Toyoshiba,
H./Gaiteri, C./Chaibub Neto, E. (2015). Bayesian network reconstruction using systems
genetics data: Comparison of MCMC methods. Genetics 199 (4), 973–989. https://doi.org/10.1534/genetics.114.172619.
Tibshirani, R. (1996). Regression shrinkage and
selection via the lasso. Journal of the Royal Statistical Society: Series B
(Methodological) 58 (1), 267–288. https://doi.org/10.1111/j.2517-6161.1996.tb02080.x.
Wu, S.-H./Chen, K.-L./Hsu, C./Chen, H.-C./Chen,
J.-Y./Yu, S.-Y./Shiu, Y. (2022). Creatine supplementation for muscle growth: A scoping
review of randomized clinical trials from 2012 to 2021. Nutrients 14 (6). https://doi.org/10.3390/nu14061255.
Zhao, T./Liu, H./Roeder, K./Lafferty,
J./Wasserman, L. (2020). The huge package for high-dimensional undirected graph
estimation in R.