The St. Nicolas House algorithm (SNHA) finds association chains of direct
dependent variables in a data set. The dependency is based on the correlation
coefficient, which is visualized as an undirected graph. The network prediction
is improved by a bootstrap routine. It enables the computation of the empirical
p-value, which is used to evaluate the significance of the
predicted edges. Synthetic data generated with the Monte Carlo method were used
to firstly compare the Python package with the original R package, and secondly
to evaluate the predicted network using the sensitivity, specificity, balanced
classification rate and the Matthew's correlation coefficient (MCC). The Python
implementation yields the same results as the R package. Hence, the algorithm
was correctly ported into Python. The SNHA scores high specificity values for
all tested graphs. For graphs with high edge densities, the other evaluation
metrics decrease due to lower sensitivity, which could be partially improved by
using bootstrap,while for graphs with low edge densities the algorithm achieves
high evaluation scores. The empirical p-values indicated that the predicted
edges indeed are significant.
Keywords: Python, correlation, network reconstruction, bootstrap, St. Nicolas house algorithm
Conflict of interest: There are not conflicts of interest.
Citation: Hake, T. / Bodenberger, B. / Groth , D. (2023). In Python available: St. Nicolas House Algorithm (SNHA) with
bootstrap support for improved performance in dense networks. Human Biology and Public Health 1. https://doi.org/10.52905/hbph2023.1.63.
Copyright: This is an open access article distributed under the terms of the Creative Commons Attribution License which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
The St. Nicolas house algorithm to analyse interacting, correlated variables is
now available in R and in Python. The added bootstrap routine also improves the
sensitivity of detecting associations between variables without a loss in
specificity.
Contents
Introduction
A major task in a variety of scientific fields is to identify direct interactions
between variables, as this grants insights into their underlying relationship and
possibly exposes unwanted effects, for instance in the area of medicine. These
interactions can be used to obtain a network representation of the data where edges
represent the interaction with nodes, which corresponds to the data variables.
Methods such as Lasso- or Ridge-regression heavily rely on the input parameter
selection to provide adequate results. Others, like Principal component, factor or
cluster analysis are often used to visualize dependencies between the variables in a
data set, but they struggle to classify dependent and independent variables. Another
recently introduced method is the St. Nicolas house algorithm (SNHA) (Groth et al. 2019; Hermanussen et al. 2021). It is currently available in the R
package asg. To attract a wider community, the core functionality of the asg package
was reimplemented in Python (van Rossum and Drake
2009). Python was the choice, because it is currently one of the most
popular programming languages (University of
California 2022; Carbonnelle
2022).
However, methods such as simple correlation or mutual information using simple
thresholds fall short of detecting only true direct interactions, as they do not
account for indirect or transitive associations between interacting variables.
Consider, for example, a gene A directly controls a second gene B, which in turn
directly controls a third gene C. Simple correlation would falsely predict a
connection between gene A and gene C. Thus, in networks inferred from biological
data, with methods such as simple correlation, mutual information, or distance
correlation, many erroneous edges would be expected (Marbach et al. 2010; Marbach et al. 2012; Dunn et al.
2008; Burger and Nimwegen 2010;
Lapedes et al. 1997). To address this,
several methods like partial correlation (La
Fuente et al. 2005; Hemelrijk
1990; Veiga et al. 2007) and
probabilistic approaches such as maximum entropy model (Lapedes et al. 1997; Marks
et al. 2011; Hopf et al. 2012)
as well as network deconvolution (Feizi et al.
2013) are used to strengthen direct associations, while removing indirect
or transitive ones. These approaches have in common that the result is sensitive to
the selection of input parameters.
The SNHA (Groth et al. 2019; Hermanussen et al. 2021) is a parameter free
approach, which finds direct interactions between variables. The SNHA ranks the
absolute correlation coefficients in descending order and thereby creates
hierarchic, so called,association chains. Association chains are characterized by
sequences for which a reversing start and end point does not change the order of the
elements (compare Figure 1). These sequences
are used to visualize the dependencies of the underlying variables as a graph by
connecting them via undirected edges.
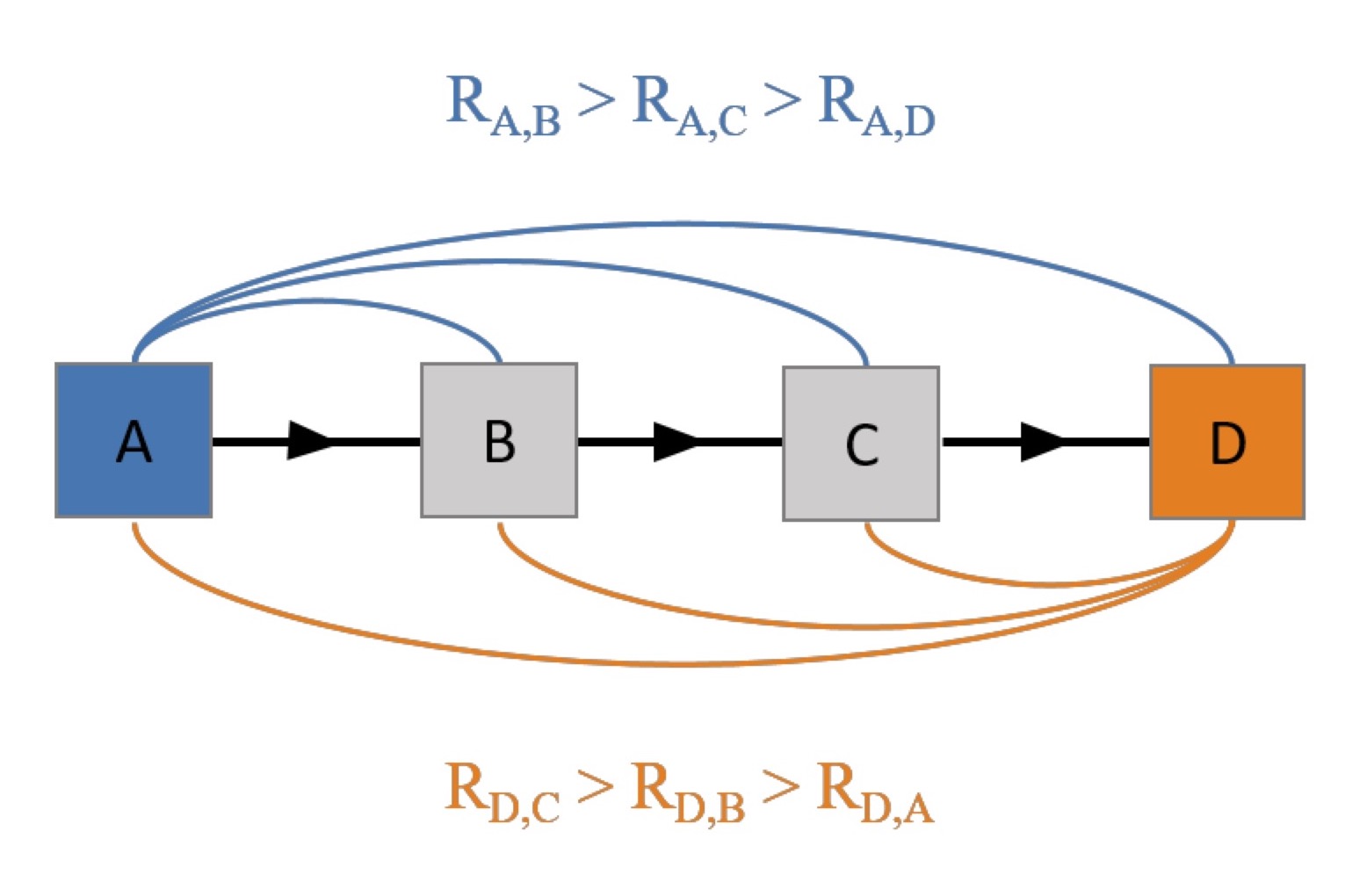
Figure 1 An exemplary association chain with four nodes. It is characterized by
the order of pairwise correlation coefficients between the node D and all
other nodes, which matches the order of the pairwise correlation
coefficients between the A and all other nodes. This ordering builds an
association chain and undirected edges connects its members.
Bootstrapping is a method that uses sampling with replacement to provide statistical
inferences such as error and bias estimates, confidence intervals, and hypothesis
tests without assumptions such as normal distributions or equal variances (Hesterberg 2011). Colby et al. (Colby et al. 2018) have shown that bootstrap
aggregation in inferred networks improves stability and, depending on the size of
the input data set, it might increases the accuracy. Further, applying bootstrapping
to biological networks like gene regulatory networks, can identify high-confidence
edges and well-connected hub nodes that potentially play important roles in
understanding the underlying biological processes of these networks(Li et al. 2011). The aim of this work is to
obtain empirical p-values for predicted edges using the
bootstrapping approach, which also stabilizes the prediction quality of the
algorithm. Further, testing the algorithm on different graph types will reveal
possible limits of the algorithm. And finally, the performance of the SNHA will be
compared to the R package asg, to ensure a correct Python port.
Materials and Methods
Software
Major functionalities of the R packages asg (Groth et al. 2019; Hermanussen
et al. 2021) and mcgraph (Novine
et al. 2022) for the open source software R (R Core Team 2022) were reimplemented using the
programming language Python 3 (van Rossum and
Drake 2009) (version 3.8.10). Here, the core packages used are pandas
(1.3.4), numpy (1.21.3), igraph (0.9.11) and matplotlib (3.4.3). The current R
version is available at Groth (2023), while the Python version is
available at Hake (2023).
Data
The data used for the analysis was synthetic data for which the underlying graph
structure was known. The data was created by the Python implementation of the
function mcgraph.graph2data (Novine et al.
2022). It creates data of varying size and density, independent from
the graph type using the Monte Carlo method (Metropolis and Ulam 1949), which serves as an input for the
SNHA.
Graph types
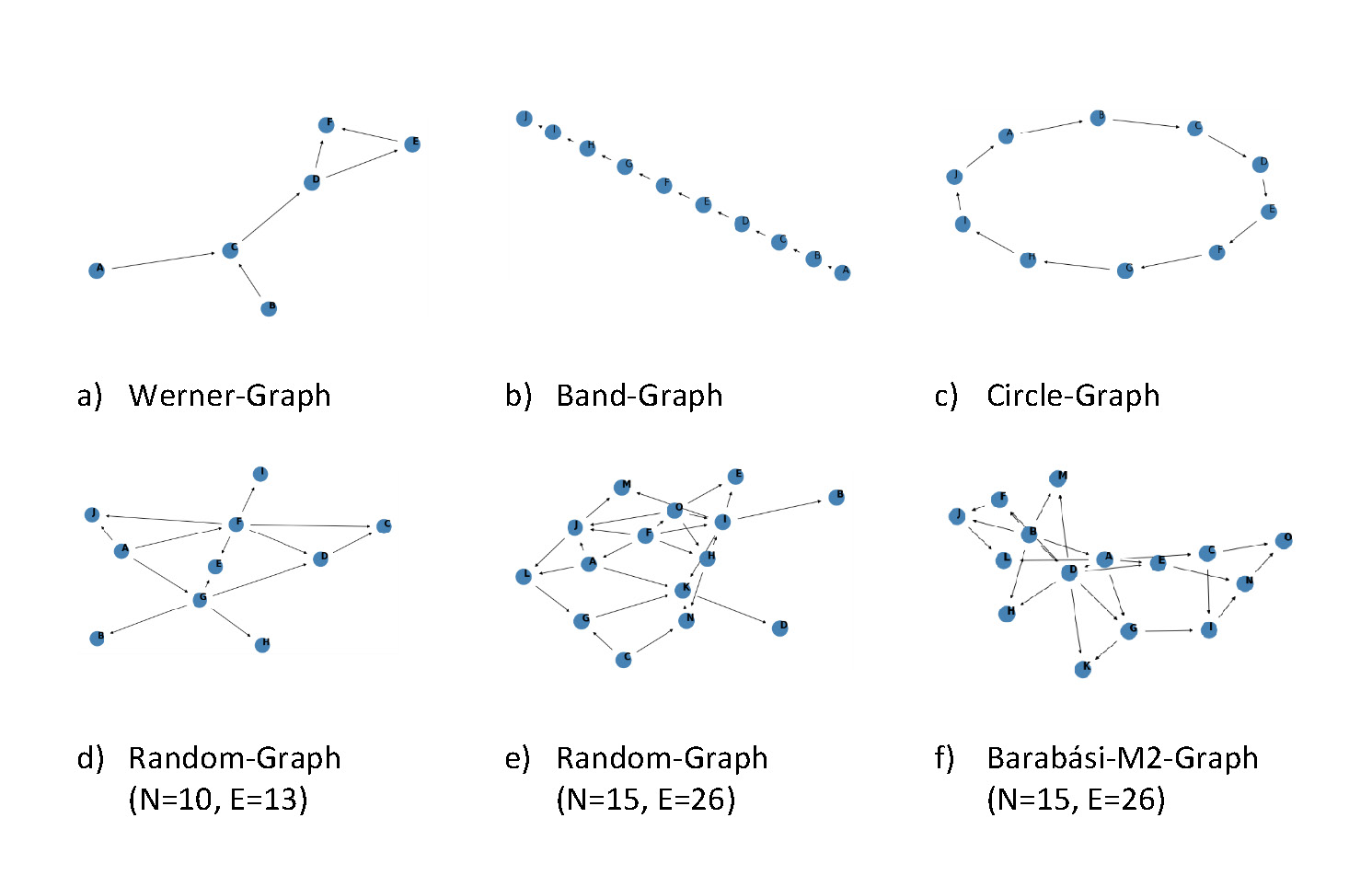
Figure 2 The different graph types used for the analysis.
In the analysis different types of graphs with a diversity in their topology and
density were used to get a better understanding in which settings the algorithm
performs well and in which not. Examined graph types were Werner-, Hub-, Band-,
Circle-, and Random-Graphs (Figure 2).
a)
Werner-Graph: An artificial graph containing the
following features: the nodes A and B converges in node C, hence
both nodes affect node C. C is also connected to node D, which
diverges to node E and F. The connection between nodes E and F
creates a cycle of the nodes D, E and F.
b)
Band-Graphs: These are connected graphs that are
characterized by each node of the graph having a degree of two and
two nodes having a degree of one.
c)
Circle-Graphs: Similar to the Band-Graph, but the last
node is also connected to the first node.
d)
Random-Graphs: Randomly assign undirected edges between
the nodes. Afterwards, randomly select starting nodes. Draw an edge
from each starting point to its neighbours. Start from each
neighbour until all nodes are visited once. These random graphs were
compared to the scale-free Barabási graphs e), f)
(Barabási and Albert
1999).
St. Nicolas house algorithm (SNHA)
The SNHA (Groth et al. 2019; Hermanussen et al. 2021) takes a
correlation matrix as its input. In the first step it then loops over the
columns of the correlation matrix where each column corresponds to a node in the
graph. For each column, the correlations are first ordered according to their
absolute values. Then, it looks for association chains by checking if the
ordered correlations match forward and backward. If that is the case, a
so-called direct chain is found. In case a direct chain was not found in the
above step, the algorithm looks for middle chains. In this case, the examined
node is placed in all possible positions in the middle of the chain, and it is
checked if the resulting sequence of nodes has an ordering of correlation
coefficients that matches from forward/backward. After the algorithm has
searched for middle chains, the row and the column with the lowest correlated
value to the node that is examined at the moment is removed from the correlation
matrix and the algorithm continues to search for a direct chain. This procedure
is followed until either a chain was found or only 3 nodes remain for the
examined chain. After that, the next node is examined. Found chains are then
used to establish an undirected graph by establishing undirected edges between
the nodes along detected chains.
Algorithm 1 Saint Nicolas House Algorithm
input data;
procedure Saint_Nicolas_House Analysis (input data);
matrix = correlation(input_data);
for each column of matrix do
correlation_matrix = matrix
while length(column) > 3 do
find longest chain where ordering of correlation
matches forward and backward;
if no chain was found then
search Middle-Chain;
end
remove row and column of correlation matrix with smallest
correlation coefficient to the column (node) examined at the moment;
endend
Quality measure for prediction
The metrics used to evaluate the prediction of the edges based on the
correlations is the Balanced Classification Rate (BCR) (Brodersen et al. 2010) and the Matthews correlation
coefficient (MCC) (Matthews 1975).
These measures result from the counts of true positive (TP), false positive
(FP), true negative (TN) and false negative (FN), which are gathered in a
confusion matrix. The BCR is defined as:
with
It is used to evaluate binary classification problems. Here, we predict whether
two nodes are connected by an edge or not. It is particular useful for
imbalanced data, e.g. sparse or dense adjacency matrix, which results in a graph
with few edges or a highly connected graph, respectively. The BCR is defined in
an interval of [0, 1]. An algorithm that guesses randomly should have a BCR
value of around 0.5 or slightly higher. For example, predicting all possible
edges as existing would have a sensitivity of 1, but a low specificity if the
number of real edges is low in comparison to the total number of possible edges.
The MCC is defined as:
The MCC is defined in the interval of [−1, 1], so random guesses lead to values
around zero,while solely false predictions produce values down to -1 and solely
true predictions cause values of 1.
Empirical p-values by bootstrapping
The empirical p-values (Davison
and Hinkley 1997) were calculated with the bootstrapping approach
that was applied to the synthetic data produced for the graph types described
above. Therefore, a randomized dataset is created by taking k samples (with
replacement) from the synthetic data and shuffle it column wise. Afterwards, the
edges are predicted by the SNHA. This procedure is repeated m-times and the
number of predictions per edge is counted. Repeating this procedure n-times
leads to a distribution for each edge, which is used to compute the p-value.
Also, the SNHA predicts the edges on the synthetic data m times and sums the
number of predictions per edge. The p-value is calculated by North (North et al. 2003):
r is the number of predictions of the randomized data, which is equal or greater
than the number of predictions of the synthetic data. While n is the number of
repetitions described above. On the other hand, the upper boundary of the
confidence interval of the distribution yields a significance threshold for the
number of predictions.
Results
After the implementation of the SNHA in Python it was compared to the original R
package. Therefore, the graphs from above where used to generate synthetic data
(Novine et al. 2022). This data was
passed into the R and Python packages to predict the networks. Both implementations
of the algorithm yield the same graph predictions. However, the bootstrapping
routine leads to slight differences as they depend on the resampling of the data.
The computation of the empirical p-values (Davison and Hinkley 1997) via bootstrapping demands
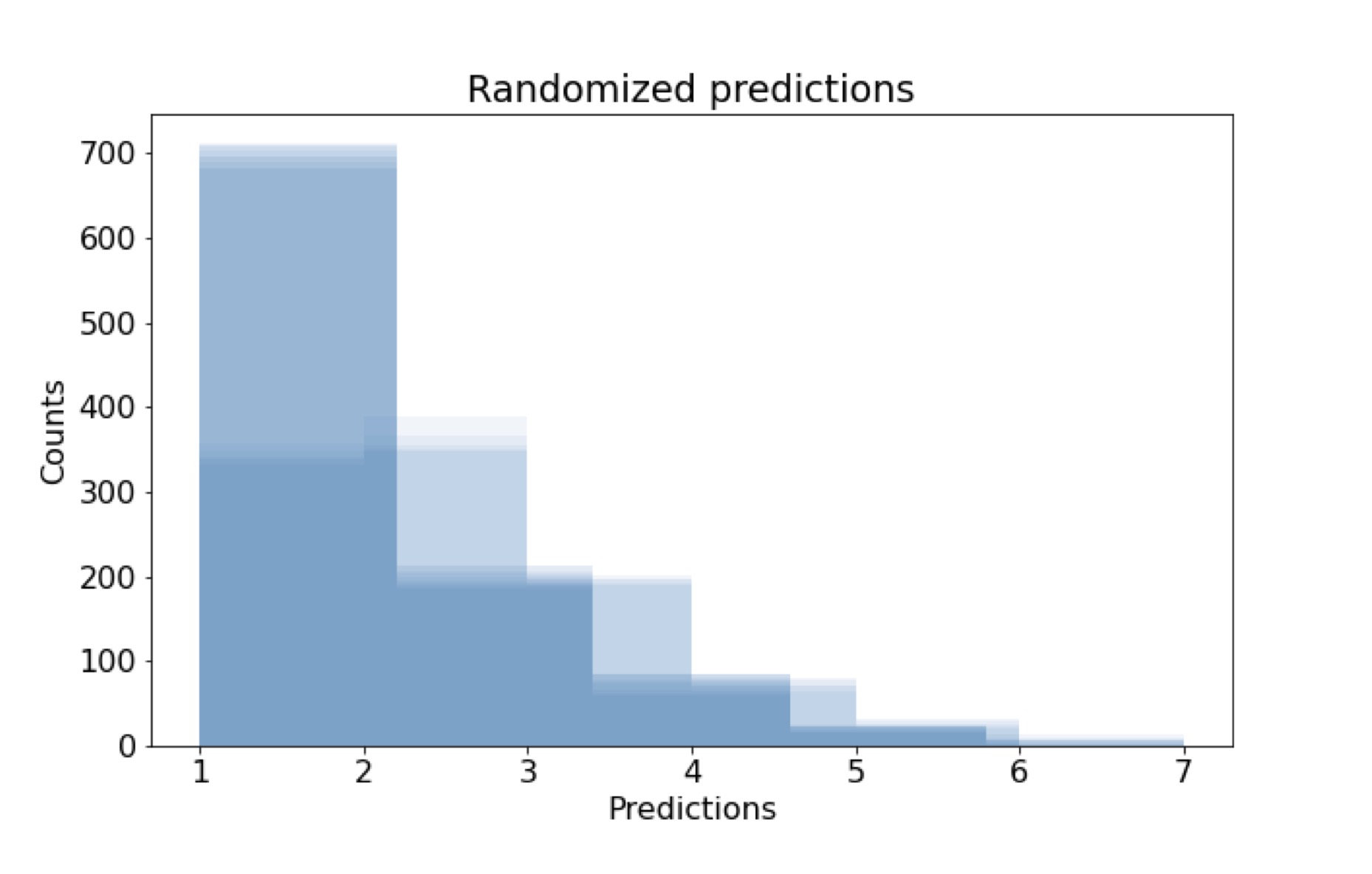
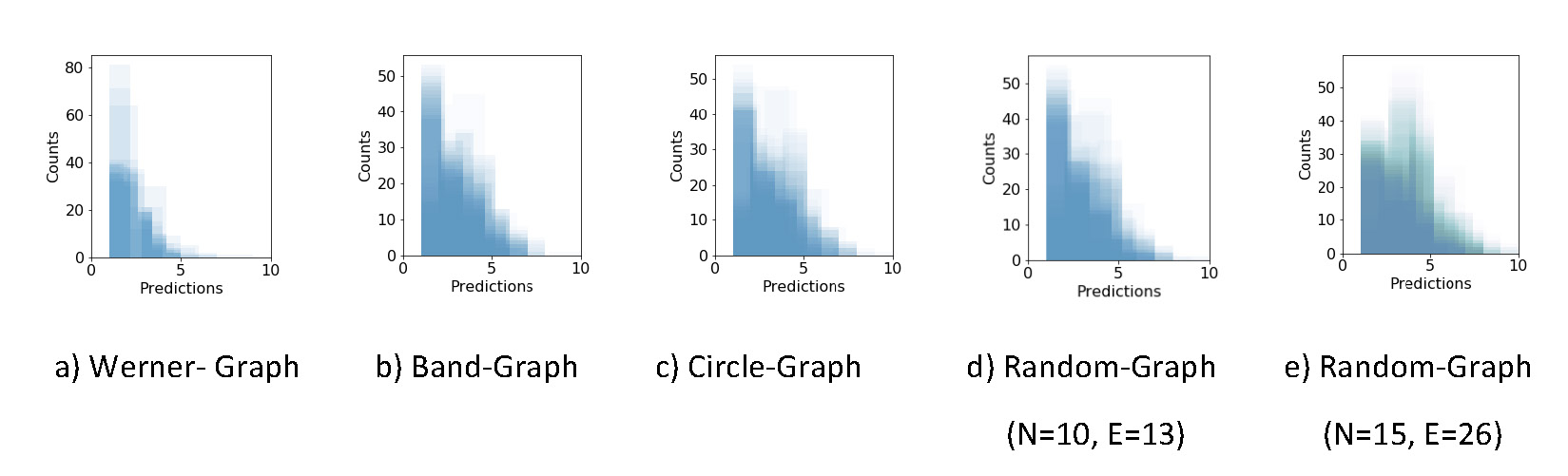
predictions on randomized data. As a test case, the Werner graph (Fig. 2a) with 1000 iterations and 100 sampling
trials per iteration was used to count the random predictions for each edge (Fig.
3). Further, the prediction on the
synthetic data without randomization is needed to compute the empirical
p-value (Eq. 1).
Figure 3 The distribution of all edge predictions after a column wise
randomization of the Werner graph. 1000 iterations with 100 sampling trials
per iteration are used to count the number of predictions. On the x-axis the
number of predictions in a single iteration (e.g., the bar height of around
100 at 4 predictions means, that in 100/1000 iterations an edge was
predicted 4 times during the 100 sampling trails).
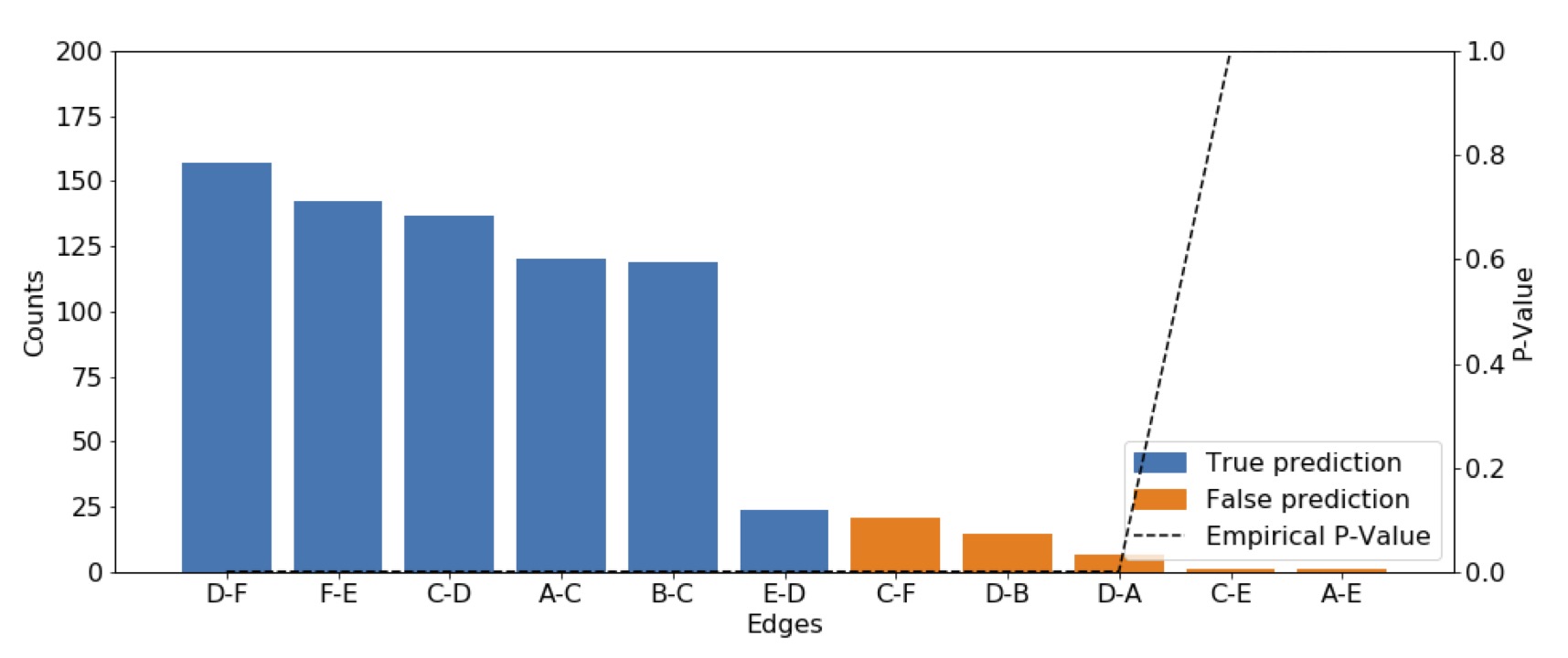
Figure 4 shows the prediction on the synthetic
data. The blue bars represent the cumulative count of the true predictions, while
the orange bars account for the false predictions in the Werner graph (Fig. 2a). Next to the number of predictions the
empirical p-value for each edge is plotted, which is below the
significance threshold of 0.05 for the edges D-F, F-E, C-D, A-C, B-C, D-E, C-F, D-B,
and D-A (Fig. 2 a).
As the number of predictions drops towards one the p-value grows to one. In order to
reduce the computational effort to compute the empirical p-values,
assume the randomized predictions are binomial distributed. Then the
p-value computation reduces from the multiple prediction of
random data to a binomial test with a certain probability of success. The test
statistic z on the null hypothesis is the following:
and
with
is the probability of success proposed by the
distribution of predictions on the random data, is computed by sampling from a binomial
distribution and n1 = n2 = 1000 are the
number of iterations. The null hypothesis will be rejected if |z| > 1.65, which
is the critical value for a right sided z-test (the limitations for the test are
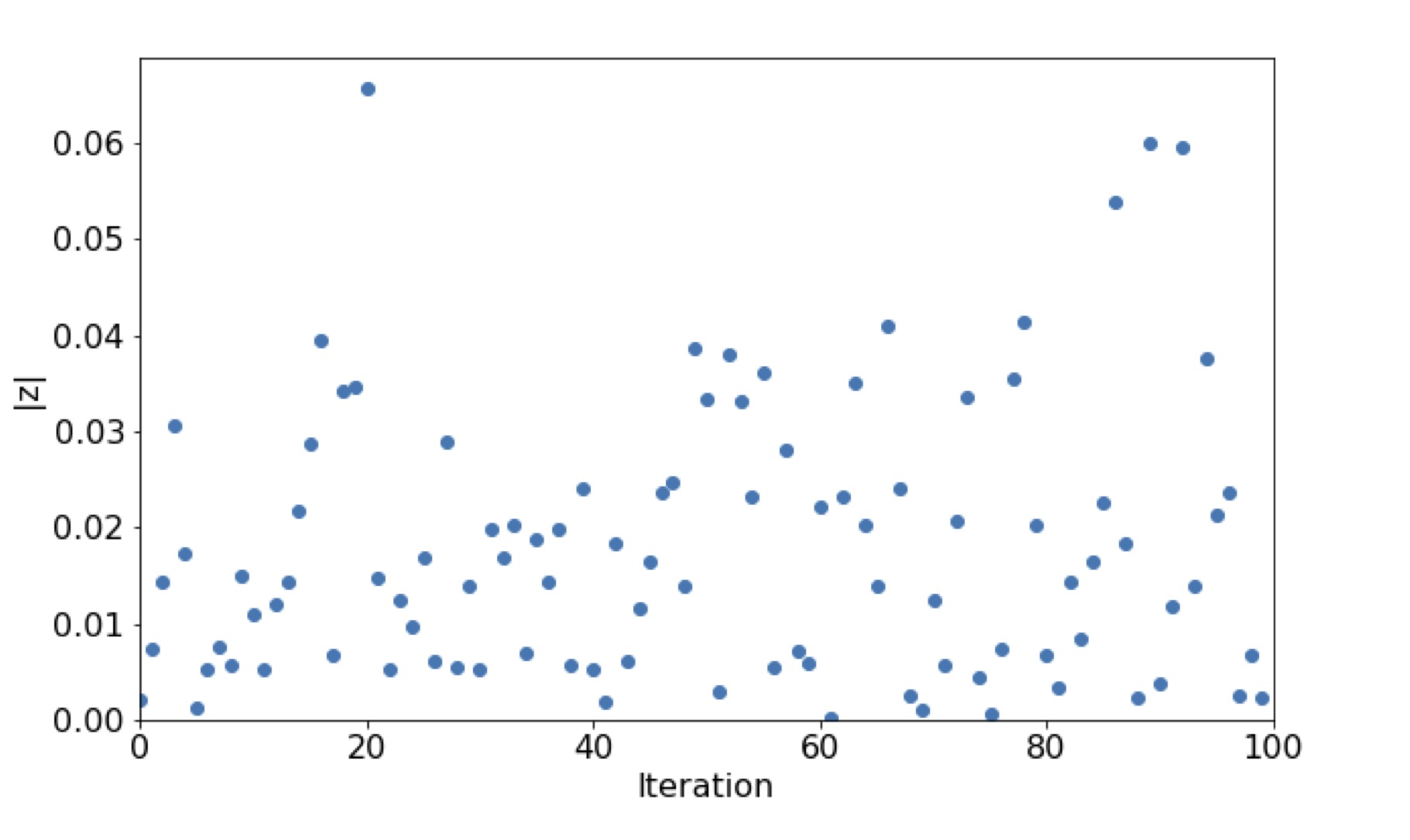
discussed later). Figure 5 shows 100
repetitions for the z calculation for the proposed computed from the distribution in figure 3. The null hypothesis holds true (Fig. 5), so the distribution of the predictions on
the randomized data follows the binomial distribution with a success probability of
.
Figure 4 Cumulative predictions of undirected edges in the Werner graph on 100
samples. The true predictions are plotted in blue, while the false
predictions are plotted in orange. The predictions are undirected so the
maximum appearance of an edge is 200. The black dotted line is the empirical
p-value (e.g., for the edge C-E the p-value is 1.0).
Figure 5 The absolute value of the right
sided z-test statistic. The null hypothesis H0: p1 = p2 will be rejected if
|z| > 1.65 (Eq. 2). p1 is the probability of success estimated from the
distribution shown in figure 3, while p2 is estimated by sampling from a
binomial distribution , with n1=1000.
After showing for the test case that the distribution in figure 3 follows a binomial distribution, it can be assumed that the
distributions of the predictions on randomized data for the other graph types also
follow a binomial distribution (Fig. 8
supplement material). Now, the interest shifts to the probability of success, as the
binomial test needs this probability as an input. Here, success represents finding
an edge.
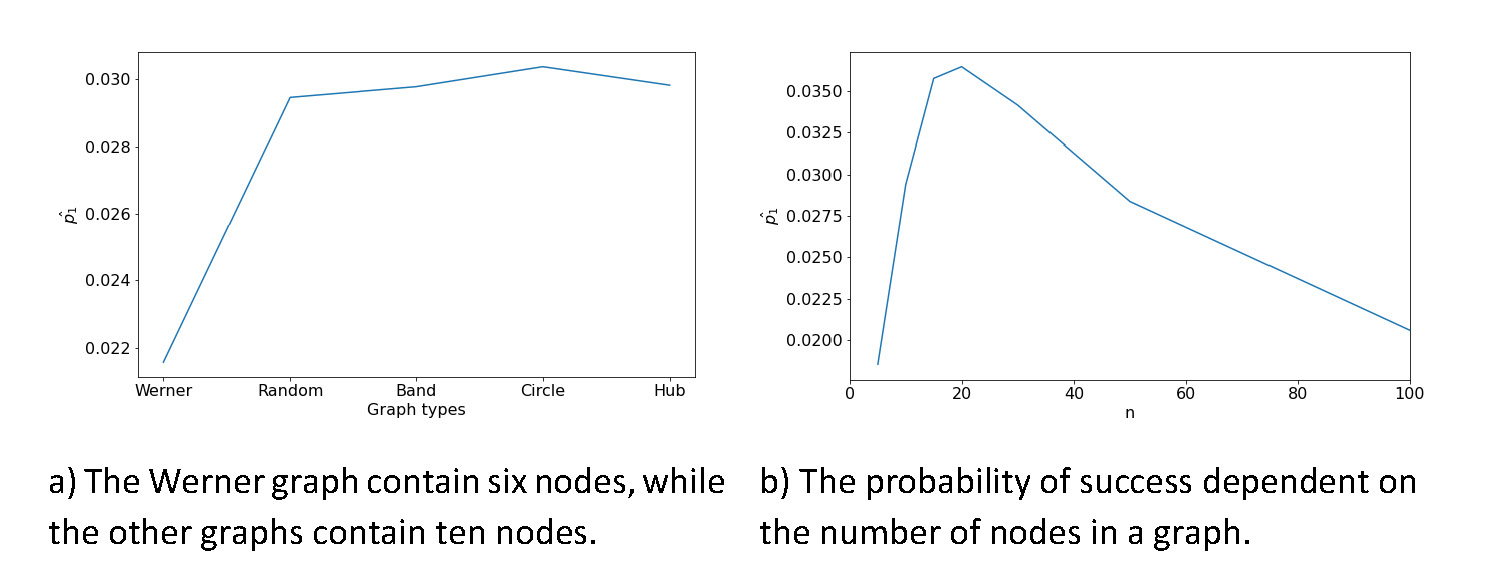
Figure 6 Influences on the probability of success, which is the likelihood of
predicting an edge. These probabilities result from the randomized data for
the different graphs (compare Fig. 8
and Eq.1).
The probability of success resulting from the distributions of the other graph types
is shown in figure 6a. The probability of success is lowest for the Werner graph and
stays constant afterwards. The main differences between the Werner graph and the
other graph types are the number of nodes. The Werner graph has six nodes, while the
other graphs in this comparison contain ten nodes. The driving factor for the
probability of success seems to be the number of nodes in a graph. To predict the
probability of success based on the number of nodes, different random graphs with 5,
10, 15, 20, 30, 50 and 100 nodes were created to compute the probability of success
(Fig. 6b). After showing that the randomized
data is distributed binomially and estimating the probability of success
(Fig. 6b),
the p-values can be computed by the binomial test. For example,
using the bootstrap settings of λ = 0.20 and n = 20 yields a
p-value ≤ 0.004 for all edges found (using
; Fig. 6b).
In general, the p-values for all edge predictions can be computed
by the binomial test. Using this test to compute the p-values
reduces the computational effort, because the number of iterations within the
bootstrap routine are reduced and the computed p-values are still
reliable.
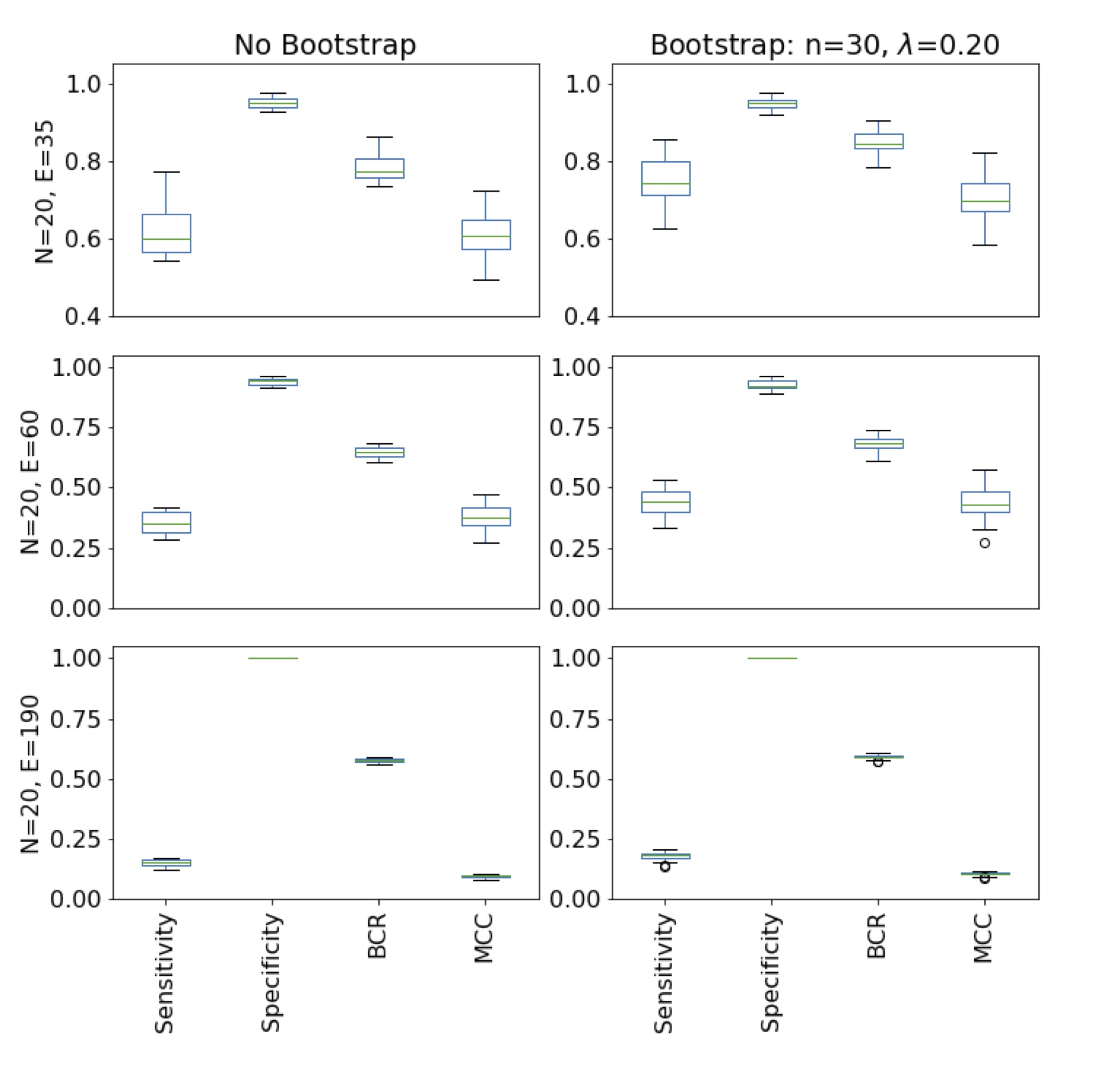
In order to compare the algorithm with and without using bootstrap 20 random graphs
(N=20, E=35 & N=20, E=60) were created. The algorithm predicts the network on
the correlation data with and without using bootstrap (Fig. 7). For this test the number of iterations within the
bootstrap routine is 30 (n=30) and the threshold for an edge to be accepted as a
prediction is 20% (λ = 0.20). In general, the statistics are similar comparing the
prediction with and without bootstrap. However, using the algorithm with bootstrap
increases the sensitivity, BCR and MCC, especially in the case of higher edge
density (see also Tab. 1). The specificity does not increase, nor decrease using the
SNHA with bootstrap. As the edge density increases, the overall performance is
reduced, but the specificity stays close to 1.00. The speed of the algorithm was
tested on a graph with 100 nodes. It runs 1.2s 14.9ms without bootstrapping (measured using timeit
cell magic in Jupyter Notebook). While the bootstrap routine increases the runtime
by a factor of about 10n (n is the number of bootstrap iterations).
Finally, the random graphs here shown were compared to the Barabási-M1 and -M2 graphs
(Barabási and Albert 1999), as these
graphs are representative for networks occurring in social and biological processes.
Here again, 20 graphs each are created and predicted using the SNHA with and without
bootstrap. The table 1 shows that the algorithms prediction on the proposed random
graph and Barabási graphs are similar. Also, the difference between using the
algorithm with bootstrap and without bootstrap behaves equally for the different
graph types.
Table 1 Comparison between the prediction on the above-described random graph
with the prediction on the Barabási-M1 and -M2 graph. Each statistic is the
mean over the score of 20 predictions. The bootstrap ran with 30 iterations
and a threshold of λ=0.20.
Sensitivity
Specificity
MCC
Graph type/Bootstrap
No
Yes
No
Yes
No
Yes
Barabási-M1 N=20, E=19
0.94
0.99
0.97
0.97
0.84
0.85
Random N=20, E=19
0.81
0.95
0.97
0.95
0.76
0.77
Barabási-M2 N=20, E=35
0.66
0.79
0.95
0.95
0.64
0.72
Random N=20, E=35
0.62
0.75
0.95
0.95
0.61
0.70
Discussion
The SNHA is a robust, non-linear and parameter free visualization method for
multivariate data. It was developed recently and verified in a few studies (Groth et al. 2019; Dorjee et al. 2021; Hermanussen et al. 2021; Scheffler
et al. 2021). It displays associations between variables in a graph
yielding immediate insights into the principle data structure. The SNHA assesses
variable chains from three up to ten nodes, which allows an understanding of
large-scale interactions between the variables. The search for tools to reconstruct
networks based on correlations led to code snippets to identify edges. However,
these examples (Yan Holtz 2018; Cortez 2017) do not go beyond pairwise
comparisons of correlations. Since the code was only available in a R package,its
use was limited to researchers comfortable in this programming language, so, it was
ported to Python. The Python port yields the same results for the same data sets as
the R asg package.
The empirical p-value computation and the binomial test suggest that
all edge predictions occurring in 7% of the bootstrap iterations are statistically
significant. Next, the p-value computation is discussed in more detail. The
significance of the found edges are computed both empirically and by the binomial
test. In order to be statistically sound, the predictions on randomized data needs
to be binomial distributed. To test for binomial distribution the right sided z-test
was conducted, which is valid if and and similarly for and n2. On the low probability of
success (Fig. 6b) and a n = 100, the condition > 5 is not met. However, the empirical
p-values as well as the binomial test suggeststhat an edge
becomes significant if it is predicted six out of 100 times with the bootstrap
routine. Selecting all significant edges as predictions would increase the false
positive predictions. The restrictive parameter λ acts as a threshold for the number
of appearances of an edge within the bootstrap routine (e.g., λ = 0.5 means an edge
needs to be found in 50% of all bootstrap iterations to be accepted as a predicted
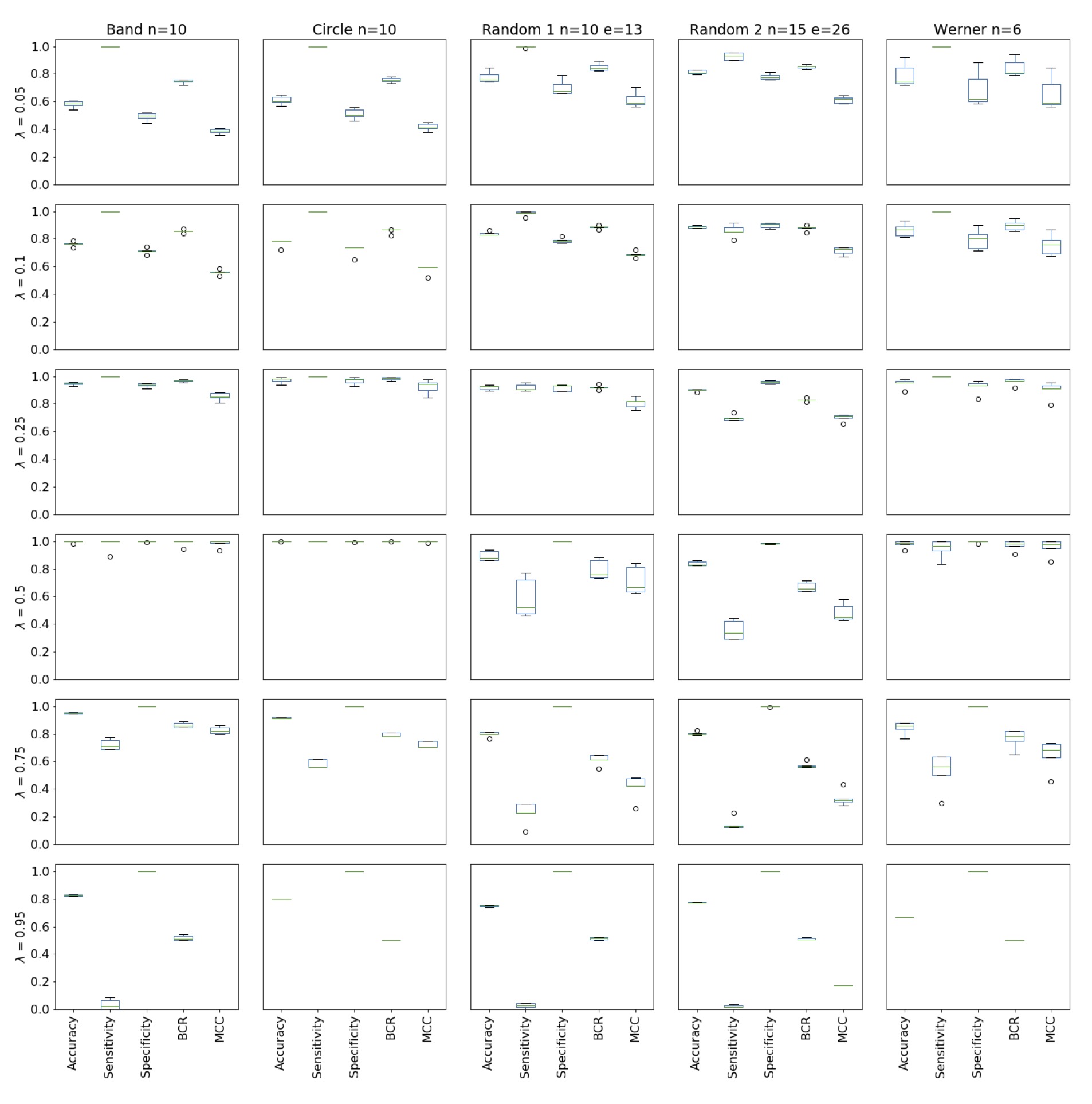
edge). The impact of the threshold on the network predictions is shown in the
supplement material (Fig. 9). It is unlikely to find an association chain randomly,
as the probability of success estimations from the distribution of number of edge
predictions on randomized data yields . In a real-world example one could not find the
optimal λ-value, as the underlying network structure is not known. So, to only
interpret highly significant edges, a λ ≥ 0.20 might be reasonable, even though the
condition of the z-test does not hold.
Figure 7 Comparison between the normal St. Nicolas house algorithm used with and
without bootstrap. The statistics are computed with the prediction of 20
different random graphs with 20 nodes and 35/60/190 edges each. The
bootstrap accepts edges as a prediction if an edge occurs 20% of the
iterations (n=30). On one hand, the prediction gets worse as the edge
density increases. On the other hand, when using the algorithm with
bootstrap the prediction improves.
The predictive power of the algorithm with and without bootstrap was assessed by the
metrics sensitivity, specificity, BCR, and MCC on graphs with 20 nodes and a
different number of edges (Fig. 7). In
general, the evaluation scores decreases as the number of edges increases,while at
low edge densities the algorithm performs well. First, the edges found with the
algorithm are real edges, as the specificity are in all cases is close to 1.00.
Second, the algorithm is better than random guessing as the BCR > 0.5 and the MCC
> 0 for all tested graph-types and edge densities. In fact, for graphs with 20
nodes and 35 edges or less it performs very well, as the statistics reaches close to
1.00 (Tab. 1, Fig. 9 in supplement material) for small graphs. Even though the
bootsrap approach maintains the pattern of reducing evaluation scores, it improves
the prediction (Fig. 7, Tab. 1). However, the slight improvement might not
justify the increase in time needed to use the bootstrap approach, as it repeats the
algorithm n times. The largest increase is measured in the sensitivity. As the
sensitivity increases, while the high specificity stays constant, the BCR and MCC
also increase. The algorithm looks for chains and it only finds those which are
distinct in correlation. Especially, those chains which contain branches will not be
mapped completely, but only one branch will be found. The bootstrap routine helps to
identify the branches of the chains as well. By sampling the data set in some cases
one branch will be more distinct, while the other branch is emphasized in another
case. So, the bootstrap routine identifies further edges which increases the true
positive rate. Applying the algorithm on the Barabási-M1 and -M2 yields similar
results as the predictions on the random graph proposed above. Therefore, these
tests enhance the described results even further. However, the performance is
slightly better on the Barabási graphs, because these graphs start with only one
controlling nodewhile the random graphs described above contain two controlling
nodes, from which the directed graph is built. The controlling points might
interfere with each other as the paths converge into a single node. This
interference reduces the degree of distinction in correlation, which complicates the
extraction of association chains.
Conclusion
The SNHA performs well on the tested graph types, especially on low edge densities.
For higher edge densities the prediction can also be improved using the bootstrap
routine. The predicted edges are statistically significant. The SNHA (Groth et al. 2019; Hermanussen et al. 2021) was successfully ported from the R
package asg to Python (Hake 2023). Both
packages yield the same network predictions for the same data sets. Now the SNHA is
available in R and Python with direct access to the algorithm for researchers
comfortable with these programming languages. Since there is no other tool that goes
beyond comparing pairwise correlations, the SNHA could be an enrichment for certain
parts of the Python community.
Appendix
Supplement material
Figure 8 The distribution of all edge predictions after a column wise
randomization of the Werner graph. 100 iterations with 100 sampling
trials per iteration are used to count the number of predictions. On
the x-axis the number of predictions in a single iteration is shown.
Figure 9 The influence of the λ threshold on the prediction value of the
St. Nicolas house algorithm with bootstrap. For each graph five data
sets were created. The algorithm predicts each network five times
and the mean over the five statistics were computed. Doing it for
all five data sets with the corresponding λ threshold leads to the
figure above. Choosing λ high (0.75, 0.95) leads to a strong
restriction and the predictions are close to a random guess. On the
other hand, choosing λ to be low (0.05, 0.10), the false positive
number increases and the predictive power becomes low. Also, the
edge predictions might not be significant. For the test graph (types
Band-, Circle-, and Werner-Graph) a threshold of 0.50 is sufficient
to achieve predictions, which are close to the input graph. In order
to get high evaluation scores on the random graphs the threshold is
0.25.
Acknowledgements
Special thanks go to PD Dr. Christiane Scheffler and Prof. Dr. Michael Hermanussen
for organising the Summer School 2022 on “Human Growth and Development” in Gülpe,
Brandenburg. There I had space to start my project, even it is not part of the focus
of the Summer School. Further, both of them provided support finishing the
project.
Brodersen, K. H./Ong, C. S./Stephan, K.
E./Buhmann, J. M. (2010). The Balanced Accuracy and Its Posterior
Distribution. In: 20th International Conference on Pattern Recognition,
3121–3124.
Burger, L./Nimwegen, E. (2010).
Disentangling Direct from Indirect Co-Evolution of Residues in Protein
Alignments. PLoS computational biology 6, e1000633. https://doi.org/10.1371/journal.pcbi.1000633.
Colby, S. M./McClure, R. S./Overall, C.
C./Renslow, R. S./McDermott, J. E. (2018). Improving network inference
algorithms using resampling methods. BMC bioinformatics 19 (1),
376.
Davison, A./Hinkley, D. (1997).
Bootstrap Methods and Their Application. Journal of the American Statistical
Association 94. https://doi.org/10.2307/1271471.
Dorjee, B./Saha, P./Sen, J. (2021).
Hierarchy of Associations Between BMI-for-Agez-Scores, Growth and Family
Social Status Among Urban Bengali Girls of Siliguri Town, West Bengal: A St.
Nicolas House Analysis. Journal of the Anthropological Survey of India 70
(2), 224–239. https://doi.org/10.1177/2277436X211043631.
Dunn, S./Wahl, L. M./Gloor, G. (2008).
Mutual Information Without the Influence of Phylogeny or Entropy
Dramatically Improves Residue Contact Prediction. Bioinformatics (Oxford,
England) 24, 333–340. https://doi.org/10.1093/bioinformatics/btm604.
Feizi, S./Marbach, D./Médard, M./Kellis,
M. (2013). Corrigendum: Network deconvolution as a general method to
distinguish direct dependencies in networks. Nature biotechnology 33.
https://doi.org/10.1038/nbt.2635.
Groth, D. (2023). snha: St. Nicolas
House Algorithm for R. R package version 0.1.3. Available online at
https://github.com/mittelmark/snha (accessed
7/5/2023).
Groth, D./Scheffler, C./Hermanussen, M.
(2019). Body height in stunted Indonesian children depends directly on
parental education and not via a nutrition mediated pathway? Evidence from
tracing association chains by St. Nicolas House Analysis. Anthropologischer
Anzeiger 76 (5), 445–451. https://doi.org/10.1127/anthranz/2019/1027.
Hake, T. (2023). Snha4py: a Python
implementation of the St. Nicholas House algorithm. Available online at
https://github.com/thake93/snha4py (accessed
2/1/2023).
Hemelrijk, C. (1990). A matrix partial
correlation test used in investigations of reciprocity and other social
interaction patterns at group level. Journal of Theoretical Biology 143,
405–420. https://doi.org/10.1016/S0022-5193(05)80036-0.
Hermanussen, M./Aßmann, C./Groth, D.
(2021). Chain Reversion for Detecting Associations in Interacting
Variables—St. Nicolas House Analysis. International Journal of Environmental
Research and Public Health 18 (4). https://doi.org/10.3390/ijerph18041741.
Hopf, T./Colwell, L./Sheridan, R./Rost,
B./Sander, C./Marks, D. (2012). Three-Dimensional Structures of Membrane
Proteins from Genomic Sequencing. Cell 149, 1607–1621. https://doi.org/10.1016/j.cell.2012.04.012.
La Fuente, A. de/Bing, N./Hoeschele,
I./Mendes, P. (2005). Discovery of Meaningful Associations in Genomic Data
Using Partial Correlation Coefficients. Bioinformatics (Oxford, England) 20,
3565–3574. https://doi.org/10.1093/bioinformatics/bth445.
Lapedes, A./Giraud, B./Liu, L./Stormo,
G. (1997). Correlated Mutations in Protein Sequences: Phylogenetic and
Structural Effects. Santa Fe Institute, Working Papers 33. https://doi.org/10.1214/lnms/1215455556.
Li, S./Hsu, L./Peng, J./Wang, P. (2011).
Bootstrap inference for network construction with an application to a breast
cancer microarray study. The Annals of Applied Statistics 7. https://doi.org/10.1214/12-AOAS589.
Marbach, D./Costello, J./Küffner,
R./Vega, N./Prill, R./Camacho, D./Allison, K./Aderhold, A./Bonneau, R./Chen,
Y./Collins, J./Cordero, F./Crane, M./Dondelinger, F./Drton, M./Esposito,
R./Foygel, R./La Fuente, A. de/Gertheiss, J./Zimmer, R. (2012). Wisdom of
crowds for robust gene network inference. Nature Methods 9, 796–804.
https://doi.org/10.1038/nmeth.2016.
Marbach, D./Prill, R./Schaffter,
T./Mattiussi, C./Floreano, D./Stolovitzky, G. (2010). Revealing strengths
and weaknesses of methods for gene network inference. Proceedings of the
National Academy of Sciences of the United States of America 107, 6286–6291.
https://doi.org/10.1073/pnas.0913357107.
Marks, D./Colwell, L./Sheridan, R./Hopf,
T./Pagnani, A./Zecchina, R./Sander, C. (2011). Protein 3D Structure Computed
from Evolutionary Sequence Variation. PloS one 6, e28766. https://doi.org/10.1371/journal.pone.0028766.
Matthews, B. W. (1975). Comparison of
the predicted and observed secondary structure of T4 phage lysozyme.
Biochimica et Biophysica Acta (BBA) - Protein Structure 405 (2), 442–451.
https://doi.org/10.1016/0005-2795(75)90109-9.
Metropolis, N./Ulam, S. (1949). The
Monte Carlo Method. Journal of the American Statistical Association 44
(247), 335–341. Available online at http://www.jstor.org/stable/2280232 (accessed
9/12/2022).
North, B./Curtis, D./Sham, P. (2003). A
note on calculation of empirical P values from Monte Carlo procedure.
American journal of human genetics 72, 498–499. https://doi.org/10.1086/346173.
Novine, M./Mattsson, C. C./Groth, D.
(2022). Network reconstruction based on synthetic data generated by a Monte
Carlo approach. Human Biology and Public Health 3. https://doi.org/10.52905/hbph2021.3.26.
R Core Team (2022). R: A Language and
Environment for Statistical Computing. Vienna, Austria 2022. Available
online at https://www.R-project.org/.
Scheffler, C./Nguyen, T. H./Hermanussen,
M. (2021). Vietnamese migrants are as tall as they want to be. Human Biology
and Public Health 2. https://doi.org/10.52905/hbph.v2.12.